Packaging and storage—
Preserve in tight, light-resistant containers, under an inert gas or with a minimum of headspace.
Labeling* —
The label states that the product is Oil- and Water-Soluble Vitamins with Minerals Oral Solution. The label states the quantity of each vitamin and mineral in a given volume of the Oral Solution and, where necessary, the chemical form in which a vitamin is present, and states also the salt form of the mineral used as the source of each element. Where the product contains vitamin E, the label indicates whether it is the
d- or
dl- form. Where the product is labeled to contain panthenol, the label states the equivalent content of dexpanthenol. Where more than one
Assay method is given for a particular vitamin or mineral the labeling states with which
Assay method the product complies only if
Method 1 is not used.
Microbial enumeration  2021
2021 —
—
The total aerobic microbial count does not exceed 3000 cfu per mL, and the combined molds and yeasts count does not exceed 300 cfu per mL. The Oral Solution also meets the requirements of the tests for absence of
Salmonella species,
Escherichia coli, and
Staphylococcus aureus.
 NOTE—
NOTE—
In the following
Assays, where more than one
Assay method is given for an individual ingredient, the requirements may be met by following any one of the specified methods, the method used being stated in the labeling only if
Method 1 is not used.
Assay for vitamin A—
[NOTE—Use low-actinic glassware throughout this procedure.
]
Diluting solution—
Prepare a mixture of tetrahydrofuran and acetonitrile (1:1), and mix.
Mobile phase—
Prepare a filtered and degassed mixture of methanol, acetonitrile, and
n-hexane (46.5:46.5:7.0). Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard preparation—
[NOTE—USP Vitamin A RS is all-
trans retinyl acetate. Use it to analyze Oral Solution that contains vitamin A as retinol, retinyl acetate, or retinyl palmitate.
] Quantitatively dissolve an accurately weighed quantity of
USP Vitamin A RS in
Diluting solution to obtain a solution having a known concentration of about 0.33 mg of
USP Vitamin A RS per mL.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 3.3 mg of vitamin A to a 500-mL separatory funnel containing 10 mL of water and 20 mL of dehydrated alcohol. Add 150 mL of solvent hexane, insert the stopper, and shake for 1 minute. Add another 150 mL of solvent hexane, insert the stopper, shake, and allow the layers to separate. Discard the aqueous layer, and filter the solvent hexane extract through anhydrous sodium sulfate into a 500-mL round-bottom flask. Evaporate the solution to dryness with the aid of a rotary evaporator over a water bath maintained at about 65
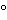
. Immediately add 10.0 mL of
Diluting solution, swirl to dissolve the residue, and filter.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 265-nm detector and a 4.6-mm × 50-cm column (prepared from two concatenated 4.6-mm × 25-cm columns) that contains packing L1. The column temperature is maintained at about 40
, and the flow rate is about 1.5 mL per minute. Chromatograph the
Standard preparation, and record the peak areas as directed for
Procedure: the relative standard deviation for replicate injections is not more than 5.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas. Calculate the quantity, in mg, of vitamin A as the retinol equivalent (C
20H
30O) in each mL of the Oral Solution taken by the formula:
10(0.872C/V)(rU / rS),
in which 0.872 is the factor used to convert retinyl acetate, obtained from
USP Vitamin A RS, to its retinol equivalent;
C is the concentration, in mg per mL, of
USP Vitamin A RS in the
Standard preparation; V is the volume, in mL, of Oral Solution taken for the
Assay preparation; and
rU and
rS are the peak areas for retinol or retinyl ester obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for cholecalciferol or ergocalciferol (vitamin D)—
[NOTE—Where vitamin D (cholecalciferol or ergocalciferol) is specified in the following procedure, use the chemical form present in the formulation and the relevant USP Reference Standard. Use low-actinic glassware throughout this procedure.
]
Diluting solution and Mobile phase—
Prepare as directed in the Assay for vitamin A.
Standard preparation—
Dissolve an accurately weighed quantity of
USP Cholecalciferol RS or
USP Ergocalciferol RS in
Diluting solution, and quantitatively dilute with
Diluting solution to obtain a solution having a known concentration of about 5 µg per mL.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 50 µg of cholecalciferol or ergocalciferol, to a 500-mL separatory funnel containing 10 mL of water and 20 mL of dehydrated alcohol. Add 150.0 mL of solvent hexane, insert the stopper, and shake for 1 minute. Add another 150 mL of solvent hexane, insert the stopper, shake, and allow the layers to separate. Discard the aqueous layer. Drain the solvent hexane extract through anhydrous sodium sulfate into a 500-mL round-bottom flask. Evaporate the solution to dryness with the aid of a rotary evaporator over a water bath maintained at about 65
. Immediately add 10.0 mL of
Diluting solution, swirl to dissolve the residue, and filter.
Chromatographic system (see Chromatography 621)—
Proceed as directed for
Chromatographic system in the
Assay for vitamin A. Chromatograph the
Standard preparation, and record the peak heights as directed for
Procedure: the relative standard deviation for replicate injections is not more than 5.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak heights for vitamin D. Calculate the quantity, in µg, of cholecalciferol (C
27H
44O) or of ergocalciferol (C
28H
44O) in each mL of the Oral Solution taken by the formula:
1.09(10C/V)(rU / rS),
in which 1.09 is a correction factor to account for the average amount of previtamin D in the Oral Solution;
C is the concentration, in µg per mL, of
USP Cholecalciferol RS or
USP Ergocalciferol RS in the
Standard preparation; V is the volume, in mL, of Oral Solution taken for the
Assay preparation; and
rU and
rS are the vitamin D peak heights obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for vitamin E—
[NOTE—Use low-actinic glassware throughout this procedure.
]
Diluting solution—
Prepare a mixture of acetonitrile and ethyl acetate (1:1).
Potassium hydroxide solution—
Transfer 90 g of potassium hydroxide pellets to a 100-mL volumetric flask containing about 60 mL of water. Mix to dissolve, cool, and dilute with water to volume.
Mobile phase—
Prepare a filtered and degassed mixture of methanol, acetonitrile, and
n-hexane (46.5:46.5:7.0). Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard preparation—
Dissolve an accurately weighed quantity of
USP Alpha Tocopherol RS in
Diluting solution, and dilute quantitatively, and stepwise if necessary, to obtain a solution having a known concentration of about 0.3 mg per mL.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 1.5 mg of alpha tocopherol, to a 125-mL conical flask fitted with a ground-glass joint, and add 25.0 mL of dehydrated alcohol. Attach a reflux condenser, and reflux in a boiling water bath for 1 minute. Cautiously add 3 mL of
Potassium hydroxide solution through the condenser, and continue to reflux for 30 minutes. Remove the flask from the bath, and rinse the condenser with about 15 mL of water. Cool, and transfer with a minimum volume of water to a 250-mL separatory funnel. Rinse the flask with 50 mL of
n-hexane, and add the rinsings to the separatory funnel. Insert the stopper, shake vigorously for 1 minute, and allow the layers to separate. Drain the aqueous layer into a second 250-mL separatory funnel, and repeat the extraction with 50 mL of
n-hexane. Discard the aqueous layer, and combine the hexane extracts. Wash the combined extracts with 25 mL of water, allow the layers to separate, and discard the aqueous layer. Add 3 drops of glacial acetic acid, and repeat the washing procedure two more times. Filter the washed hexane layer through anhydrous sodium sulfate into a 250-mL round-bottom flask. Rinse the funnel and sodium sulfate with
n-hexane, and add the rinsing to the hexane solution in the flask. Evaporate the hexane solution to dryness with the aid of a rotary evaporator over a water bath maintained at about 50
. Immediately add 5.0 mL of
Diluting solution, and swirl to dissolve the residue.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 291-nm detector and a 4.6-mm × 25-cm column that contains packing L1. The column temperature is maintained at about 40
, and the flow rate is about 3 mL per minute. Chromatograph the
Standard preparation, and record the peak heights as directed for
Procedure: the relative standard deviation for replicate injections is not more than 5%.
Procedure—
Separately inject equal volumes (about 20 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak heights. Calculate the quantity, in mg, of alpha tocopherol (C
29H
50O
2) in each mL of the Oral Solution taken by the formula:
5(C/V)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Alpha Tocopherol RS in the
Standard preparation; V is the volume, in mL, of Oral Solution taken; and
rU and
rS are the peak heights for alpha tocopherol obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for ascorbic acid, Method 2—
Transfer an accurately measured volume of Oral Solution, equivalent to about 80 mg of ascorbic acid, to a conical flask. Add 50 mL of water, 100 mL of 0.1 N sulfuric acid VS, and 15.0 mL of 0.1 N iodine VS. Stir the contents for 30 seconds, add 5 mL of starch TS, and immediately titrate with 0.1 N sodium thiosulfate VS to the disappearance of the color. Each mL of 0.1 N iodine is equivalent to 8.806 mg of C6H8O6.
Assay for calcium ascorbate, Method 2—
Proceed as directed in the Assay for ascorbic acid, Method 2. Each mL of 0.1 N iodine is equivalent to 10.66 mg of C12H14CaO12·2H2O.
Assay for sodium ascorbate, Method 2—
Proceed as directed in the Assay for ascorbic acid, Method 2. Each mL of 0.1 N iodine is equivalent to 9.905 mg of C6H7NaO6.
Assay for biotin, Method 1—
Proceed as directed in the
Assay for biotin, Method 2 under
Oil- and Water-Soluble Vitamins with Minerals Tablets, except to read Oral Solution in place of Tablets and to use the following
Assay preparation.
Assay preparation—
Transfer an accurately measured volume of Oral Solution to a volumetric flask, and dilute quantitatively, and stepwise if necessary, to obtain a solution having a concentration of 0.1 ng of biotin per mL.
Assay for biotin, Method 2—
Proceed as directed in the
Assay for biotin, Method 3 under
Oil- and Water-Soluble Vitamins with Minerals Tablets, except to read Oral Solution in place of Tablets and to use the following
Assay preparation.
Assay preparation—
Transfer an accurately measured volume of Oral Solution to a volumetric flask, and dilute quantitatively, and stepwise if necessary with water, to obtain a solution having a concentration of 0.6 µg of biotin per mL. Adjust the solution with either dilute acetic acid or 0.1 N sodium hydroxide to a pH of between 6.0 and 7.0.
Assay for cyanocobalamin—
Proceed as directed in the
Assay for cyanocobalamin, Method 2 under
Oil- and Water-Soluble Vitamins with Minerals Tablets, except to read Oral Solution in place of Tablets and to use the following
Assay preparation.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 1.0 µg of cyanocobalamin, to an appropriate vessel containing, for each mL of the Oral Solution taken, 25 mL of an aqueous extracting solution prepared just prior to use to contain, in each 100 mL, 1.29 g of dibasic sodium phosphate, 1.1 g of anhydrous citric acid, and 1.0 g of sodium metabisulfite. Autoclave the mixture at 121
for 10 minutes. Allow any undissolved particles of the extract to settle, and filter or centrifuge if necessary. Dilute an aliquot of the clear solution with water to obtain a final solution containing vitamin B
12 activity approximately equivalent to that of the
Standard preparation.
Assay for calcium pantothenate, Method 1—
Dilute phosphoric acid—
Dilute 11.8 mL of phosphoric acid with water to 100.0 mL, and mix.
Mobile phase—
Mix 30 mL of methanol with 970 mL of 0.2 M monobasic sodium phosphate. Adjust with
Dilute phosphoric acid to a pH of 3.2 ± 0.1, mix, and filter. Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard preparation—
Dissolve an accurately weighed quantity of
USP Calcium Pantothenate RS in
Mobile phase to obtain a solution having a known concentration of about 80 µg per mL.
System suitability solution—
Dissolve an accurately weighed quantity of
USP Racemic Panthenol RS in
Mobile phase to obtain a solution having a known concentration of about 80 µg per mL. Transfer 5.0 mL of this solution to a 10-mL volumetric flask, dilute with
Standard preparation to volume, and mix.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with Mobile phase to obtain a solution having a concentration of about 80 µg of calcium pantothenate per mL.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 210-nm detector and a 4.0-mm × 10-cm column that contains packing L1. The flow rate is about 1.0 mL per minute. Chromatograph 20 µL of the
System suitability solution, and record the peak areas as directed for
Procedure: the resolution,
R, between panthenol and calcium pantothenate is not less than 1.5; and the tailing factor for both the calcium pantothenate and the panthenol peaks is not more than 2.0. Chromatograph the
Standard preparation, and record the peak areas as directed for
Procedure: the relative standard deviation for replicate injections is not more than 2%.
Procedure—
Separately inject equal volumes (about 20 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas obtained for calcium pantothenate. Calculate the quantity, in mg, of calcium pantothenate (C
18H
32CaN
2O
10) in each mL of the Oral Solution taken by the formula:
C(L/D)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Calcium Pantothenate RS in the
Standard preparation; L is the labeled quantity, in mg per mL, of calcium pantothenate in the Oral Solution taken;
D is the concentration, in mg per mL, of calcium pantothenate in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution; and
rU and
rS are the peak areas for calcium pantothenate obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for calcium pantothenate, Method 2—
Proceed as directed in the
Assay for calcium pantothenate, Method 2 under
Oil- and Water-Soluble Vitamins with Minerals Tablets, except to read Oral Solution in place of Tablets and to use the following
Assay preparation.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 50 mg of calcium pantothenate, to a 1000-mL volumetric flask containing 500 mL of water. Add 10 mL of 0.2 N acetic acid and 100 mL of sodium acetate solution (1 in 60), dilute with water to volume, and filter. Dilute an accurately measured volume of this solution quantitatively, and stepwise if necessary, with water to obtain a solution having about the same concentration as that of the Standard preparation.
Assay for dexpanthenol or panthenol, Method 1—
Dilute phosphoric acid, Mobile phase, and Chromatographic system—
Proceed as directed in the Assay for calcium pantothenate, Method 1.
System suitability solution—
Dissolve an accurately weighed quantity of
USP Calcium Pantothenate RS in
Mobile phase to obtain a solution having a known concentration of about 80 µg per mL. Transfer 5.0 mL of this solution to a 10-mL volumetric flask, dilute with
Standard preparation to volume, and mix.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with Mobile phase to obtain a solution having a concentration of about 80 µg of dexpanthenol or panthenol per mL.
Procedure—
Separately inject equal volumes (about 20 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas obtained for panthenol. Calculate the quantity, in mg, of dexpanthenol (C
9H
19NO
4) or panthenol (C
9H
19NO
4) in each mL of the Oral Solution taken by the formula:
C(L/D)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Dexpanthenol RS or
USP Racemic Panthenol RS in the
Standard preparation; L is the labeled quantity, in mg per mL, of dexpanthenol or panthenol in the Oral Solution taken;
D is the concentration, in mg per mL, of dexpanthenol or panthenol in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution; and
rU and
rS are the peak areas for dexpanthenol or panthenol obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for dexpanthenol or panthenol, Method 2—
Proceed as directed in the
Assay for dexpanthenol or panthenol under
Water-Soluble Vitamins Capsules, except to read Oral Solution in place of Capsules and to use the following
Assay preparation.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 1.2 mg of dexpanthenol or 2.4 mg of panthenol, to a 100-mL volumetric flask, dissolve in and dilute with water to volume, and mix. Dilute a portion of this solution quantitatively, and stepwise if necessary, with water to obtain a solution having a concentration of about 1.2 µg of dexpanthenol or 2.4 µg of panthenol per mL.
Assay for niacin or niacinamide—
[NOTE—Use low-actinic glassware throughout this procedure.
]
Diluting solution—
Dissolve 25 g of edetate disodium in 1000 mL of water, and mix.
Mobile phase—
Mix 0.4 mL of triethylamine, 15.0 mL of glacial acetic acid, and 350 mL of methanol, and dilute with 0.008 M sodium 1-hexanesulfonate to 2000 mL. Filter, and degas. Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard preparation—
[NOTE—Use
USP Niacin RS for Oral Solution that contains niacin and
USP Niacinamide RS for Oral Solution that contains niacinamide.
] Dissolve an accurately weighed quantity of
USP Niacin RS or
USP Niacinamide RS in
Diluting solution in a volumetric flask, and dilute quantitatively, and stepwise if necessary, with
Diluting solution to obtain a solution having a known concentration of about 0.10 mg per mL.
Assay preparation—
Transfer an accurately measured volume of Oral Solution to a volumetric flask, dissolve in Diluting solution, and dilute quantitatively, and stepwise if necessary, with Diluting solution to obtain a solution having a known concentration of about 0.1 mg of niacin or niacinamide per mL.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 270-nm detector and a 4.6-mm × 25-cm column that contains packing L7. The flow rate is about 2.0 mL per minute. Chromatograph the
Standard preparation, and record the peak areas as directed for
Procedure: the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 5 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas. Calculate the quantity, in mg, of niacin (C
6H
5NO
2) or niacinamide (C
6H
6N
2O) in each mL of the Oral Solution taken by the formula:
C(L/D)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Niacin RS or
USP Niacinamide RS in the
Standard preparation; L is the labeled amount, in mg per mL, of niacin or niacinamide in the Oral Solution taken;
D is the concentration, in mg per mL, of niacin or niacinamide in the
Assay preparation, based on the labeled quantity and the extent of dilution; and
rU and
rS are the peak areas for niacin or niacinamide obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for pyridoxine hydrochloride—
Diluting solution, Mobile phase, and Chromatographic system—
Proceed as directed in the Assay for niacin or niacinamide.
Standard preparation—
Dissolve an accurately weighed quantity of
USP Pyridoxine Hydrochloride RS in
Diluting solution, and dilute quantitatively, and stepwise if necessary, with
Diluting solution to obtain a solution having a known concentration of about 0.024 mg per mL.
Assay preparation—
Transfer an accurately measured volume of Oral Solution to a volumetric flask, dissolve in Diluting solution, and dilute quantitatively, and stepwise if necessary, with Diluting solution to obtain a solution having a concentration of about 0.024 mg of pyridoxine hydrochloride per mL.
Procedure—
Separately inject equal volumes (about 5 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the areas for the major peaks. Calculate the quantity, in mg, of pyridoxine hydrochloride (C
8H
11NO
3·HCl) in each mL of the Oral Solution taken by the formula:
C(L/D)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Pyridoxine Hydrochloride RS in the
Standard preparation; L is the labeled amount, in mg per mL, of pyridoxine hydrochloride in the Oral Solution taken;
D is the concentration, in mg per mL, of pyridoxine hydrochloride in the
Assay preparation, based on the labeled quantity and the extent of dilution; and
rU and
rS are the peak areas for pyridoxine hydrochloride obtained from the
Assay preparation and
Standard preparation, respectively.
Assay for riboflavin or riboflavin-5¢-phosphate sodium, Method 1—
[NOTE—Riboflavin-5
¢-phosphate sodium is quantitated against
USP Riboflavin RS in this procedure. In the chromatogram of the
Assay preparation, the riboflavin-5
¢-phosphate peak is the only peak measured for calculation.
]
Diluting solution, Mobile phase, and Chromatographic system—
Proceed as directed in the Assay for niacin or niacinamide.
Standard preparation—
Dissolve an accurately weighed quantity of
USP Riboflavin RS in
Diluting solution, by heating if necessary, in a volumetric flask, and dilute quantitatively, and stepwise if necessary, with
Diluting solution to obtain a solution having a known concentration of about 8 µg per mL.
Assay preparation—
Transfer a volume of Oral Solution, accurately measured, to a volumetric flask, dissolve in Diluting solution, and dilute quantitatively, and stepwise if necessary, with Diluting solution to obtain a solution having a known concentration of about 8 µg of riboflavin per mL.
Procedure—
Separately inject equal volumes (about 5 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas for riboflavin and riboflavin-5
¢-phosphate. The relative retention times are about 0.18 for riboflavin-5
¢-phosphate and 1.0 for riboflavin. Calculate the quantity, in mg, of riboflavin (C
17H
20N
4O
6) in each mL of the Oral Solution taken by the formula:
1.493C(L/D)(rU / rS),
in which 1.493 is the factor for converting riboflavin-5
¢-phosphate to riboflavin;
C is the concentration, in mg per mL, of
USP Riboflavin RS in the
Standard preparation; L is the labeled amount, in mg per mL, of riboflavin in the Oral Solution taken;
D is the concentration, in mg per mL, of riboflavin in the
Assay preparation, based on the labeled quantity and the extent of dilution;
rU is the peak area for riboflavin-5
¢-phosphate obtained from the
Assay preparation; and
rS is the peak area for riboflavin obtained from the
Standard preparation.
Assay for riboflavin or riboflavin-5¢-phosphate sodium, Method 2—
[NOTE—Use low-actinic glassware throughout this procedure.
]
Solvent blank—
Transfer 1 mL of 1 N hydrochloric acid and 2 mL of 2.5 M sodium acetate to a 100-mL volumetric flask, dilute with water to volume, and mix.
Riboflavin stock solution—
Transfer about 16 mg of
USP Riboflavin RS, accurately weighed, to a 100-mL volumetric flask, dissolve in 1.0 mL of 1 N hydrochloric acid and 2.0 mL of 2.5 M sodium acetate, dilute with water to volume, and mix. Transfer 10.0 mL of this solution to a 100-mL volumetric flask, dilute with water to volume, and mix.
Standard preparation—
Transfer 5.0 mL of Riboflavin stock solution to a 500-mL volumetric flask, add 5 mL of 1 N hydrochloric acid and 10 mL of 2.5 M sodium acetate, dilute with water to volume, and mix.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 0.8 mg of riboflavin, to a 100-mL volumetric flask, dilute with water to volume, and mix. Transfer 10.0 mL of this solution to a 500-mL volumetric flask, add 5 mL of 1 N hydrochloric acid and 10 mL of 2.5 M sodium acetate, dilute with water to volume, and mix.
Procedure—
Using a spectrofluorometer that has been set to zero with the
Solvent blank, determine the maximum fluorescence intensities,
IS and
IU, of the
Standard preparation and the
Assay preparation, respectively, at an emission wavelength of about 530 nm and an excitation wavelength of about 440 nm. Calculate the quantity, in mg, of riboflavin (C
17H
20N
4O
6) in each mL of the Oral Solution taken by the formula:
C(L/D)(IU / IS),
in which
C is the concentration, in µg per mL, of
USP Riboflavin RS in the
Standard preparation; L is the labeled amount, in mg per mL, of riboflavin in the Oral Solution taken;
D is the concentration, in µg per mL, of riboflavin in the
Assay preparation, based on the labeled amount in a given volume and the extent of dilution; and
IU and
IS are the fluorescence values obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for thiamine—
Diluting solution, Mobile phase, and Chromatographic system—
Proceed as directed in the Assay for niacin or niacinamide.
Standard preparation—
Dissolve an accurately weighed quantity of
USP Thiamine Hydrochloride RS in
Diluting solution, and dilute quantitatively, and stepwise if necessary, with
Diluting solution to obtain a solution having a known concentration of about 0.024 mg of
USP Thiamine Hydrochloride RS per mL.
Assay preparation—
Dissolve an accurately measured volume of Oral Solution in Diluting solution, and dilute quantitatively, and stepwise if necessary, with Diluting solution to obtain a solution having a concentration of about 0.024 mg of thiamine hydrochloride per mL.
Procedure—
Separately inject equal volumes (about 5 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas. Calculate the quantity, in mg, of thiamine hydrochloride (C
12H
17 ClN
4OS·HCl) in each mL of the Oral Solution taken by the formula:
C(L/D)(rU / rS),
in which
C is the concentration, in mg per mL, of
USP Thiamine Hydrochloride RS in the
Standard preparation; L is the labeled amount, in mg per mL, of thiamine hydrochloride in the Oral Solution taken;
D is the concentration, in mg per mL, of thiamine hydrochloride in the
Assay preparation, based on the labeled quantity and the extent of dilution; and
rU and
rS are the peak areas for thiamine hydrochloride obtained from the
Assay preparation and
Standard preparation, respectively.
NOTE—Commercially available atomic absorption standard solutions for the minerals, where applicable, may be used where preparation of a standard stock solution is described in the following Assays. Use deionized water where water is specified. Where atomic absorption spectrophotometry is specified in the Assay, the Standard preparations and the Assay preparation may be diluted quantitatively with the solvent specified, if necessary, to yield solutions of suitable concentrations adaptable to the linear or working range of the instrument.
Assay for chromium—
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with 0.125 N hydrochloric acid to obtain a solution having a concentration of about 1 µg of chromium per mL.
Procedure—
Proceed as directed for
Procedure in the
Assay for chromium under
Oil- and Water-Soluble Vitamins with Minerals Tablets, except to calculate the quantity, in µg, of chromium (Cr) in each mL of the Oral Solution taken by the formula:
C(L/D),
in which
L is the labeled quantity, in µg per mL, of chromium in the Oral Solution; and
D is the concentration, in µg per mL, of chromium in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution.
Assay for fluoride—
[NOTE—Use plastic containers throughout this procedure.
]
Ascorbic acid solution—
Dissolve 7 g of ascorbic acid in 100 mL of water, and mix.
Mobile phase—
Prepare a filtered and degassed mixture of water, alcohol, and 1 N sulfuric acid (898:100:2). Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard stock solution—
Dissolve an accurately weighed quantity of
USP Sodium Fluoride RS in water, and dilute quantitatively, and stepwise if necessary, with water to obtain a solution having a known concentration of about 220 µg per mL. This solution contains about 100 µg of fluoride per mL.
Standard preparation—
Transfer 5.0 mL of Standard stock solution to a 100-mL volumetric flask. Add 2 mL of Ascorbic acid solution, 10 mL of alcohol, and about 70 mL of water, and mix. Adjust with 1 N sodium hydroxide to a pH of 4.25 ± 0.05. Dilute with water to volume, and mix to obtain a solution having a known concentration of about 5 µg of fluoride per mL.
Assay preparation—
Transfer an accurately measured volume of Oral Solution, equivalent to about 0.5 mg of fluoride, to a 100-mL volumetric flask. Add 1 drop of hydrochloric acid, 10 mL of alcohol, and about 75 mL of water, and mix. Adjust with 1 N sodium hydroxide to a pH of 4.25 ± 0.05. Dilute with water to volume, and mix.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a conductivity detector and a combination 4.6-mm × 3-cm guard column and 7.8-mm × 30-cm analytical column that contains packing L17. The flow rate is about 0.6 mL per minute. Chromatograph the
Standard preparation, and record the peak areas as directed for
Procedure: the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 100 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas for fluoride. Calculate the quantity, in mg, of fluoride (F) in the portion of Oral Solution taken by the formula:
100C(rU / rS),
in which
C is the concentration, in mg per mL, of fluoride in the
Standard preparation; and
rU and
rS are the peak areas for fluoride obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for iodide, Method 1—
Mobile phase—
Dissolve 5.15 g of tetrabutylammonium bromide in 320 mL of acetonitrile. Dilute with water to 2000 mL, mix, filter, and degas. Make adjustments if necessary (see
System Suitability under
Chromatography 621).
Standard stock solution—
Transfer about 0.13 g of potassium iodide, accurately weighed, to a 100-mL volumetric flask. Dissolve in and dilute with Mobile phase to volume, and mix. This solution has a concentration of about 1 mg of iodide per mL.
Standard preparation—
Dilute an accurately measured volume of the Standard stock solution with Mobile phase quantitatively, and stepwise if necessary, to obtain a solution having a known concentration of about 2.5 µg of iodide per mL.
System suitability solution—
Transfer about 0.13 g of potassium iodide and 0.5 g of potassium iodate, accurately weighed, to a 100-mL volumetric flask. Dissolve in Mobile phase, using sonication if necessary, dilute with Mobile phase to volume, and mix. Transfer 1.0 mL of this solution to a 100-mL volumetric flask, dilute with Mobile phase to volume, and mix. Transfer 25.0 mL of this solution to a 100-mL volumetric flask, dilute with Mobile phase to volume, and mix.
Assay preparation—
Dissolve an accurately measured volume of Oral Solution in Mobile phase, and dilute quantitatively, and stepwise if necessary, with Mobile phase to obtain a solution having a concentration of about 2.5 µg of iodide per mL.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 225-nm detector and a 4.6-mm × 15-cm column that contains packing L1. The flow rate is about 1.5 mL per minute. Chromatograph about 30 µL of the
System suitability solution, and record the peak areas as directed for
Procedure: the relative retention times are about 0.32 for iodate and 1.0 for iodide; and the resolution,
R, between iodate and iodide is not less than 2.5. Chromatograph the
Standard preparation, and record the peak areas as directed for
Procedure: the relative standard deviation for replicate injections is not more than 2.0% for the iodide peak.
Procedure—
Separately inject equal volumes (about 30 µL) of the
Standard preparation and the
Assay preparation into the chromatograph, record the chromatograms, and measure the peak areas for iodide. Calculate the quantity, in µg, of iodide in each mL of Oral Solution taken by the formula:
(126.9/166.0)(C)(L/D)(rU / rS),
in which 126.9 is the atomic weight of iodine; 166.0 is the molecular weight of potassium iodide;
C is the concentration, in µg per mL, of potassium iodide, calculated on the dried basis, in the
Standard preparation; L is the labeled quantity, in µg per mL, of iodide in the Oral Solution;
D is the concentration, in µg per mL, of iodide in the
Assay preparation, based on the labeled quantity and the extent of dilution; and
rU and
rS are the peak areas for iodide obtained from the
Assay preparation and the
Standard preparation, respectively.
Assay for iron—
Iron standard stock solution—
Transfer about 100 mg of iron powder, accurately weighed, to a 1000-mL volumetric flask, dissolve in 6 N hydrochloric acid, dilute with water to volume, and mix.
Standard preparations—
To separate 100-mL volumetric flasks, transfer 2.0, 4.0, 5.0, 6.0, and 8.0 mL of Iron standard stock solution. Dilute the contents of each flask with 0.125 N hydrochloric acid to volume, and mix to obtain the solutions having known concentrations of about 2.0, 4.0, 5.0, 6.0, and 8.0 µg of iron per mL.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with 0.125 N hydrochloric acid to obtain a solution having a concentration of about 6 µg of iron per mL.
Procedure—
Concomitantly determine the absorbances of the
Standard preparations and the
Assay preparation at the iron emission line at 248.3 nm with an atomic absorption spectrophotometer (see
Spectrophotometry and Light-Scattering 851) equipped with an iron hollow-cathode lamp and an air–acetylene flame using 0.125 N hydrochloric acid as the blank. Plot the absorbances of the
Standard preparations versus concentration, in µg per mL, of iron, and draw the straight line best fitting the five plotted points. From the graph so obtained, determine the concentration,
C, in µg per mL, of iron in the
Assay preparation. Calculate the quantity, in mg, of iron (Fe) in each mL of the Oral Solution taken by the formula:
C(L/D),
in which
L is the labeled quantity, in mg per mL, of iron in the Oral Solution taken; and
D is the concentration, in µg per mL, of iron in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution.
Assay for magnesium—
Magnesium standard stock solution—
Transfer about 1.00 g of magnesium ribbon, accurately weighed, to a 1000-mL volumetric flask, dissolve in 50 mL of 6 N hydrochloric acid, dilute with water to volume, and mix.
Standard preparations—
Quantitatively dilute a volume of Magnesium standard stock solution with water to obtain a standard solution having a concentration of 20 µg of magnesium per mL. To separate 100-mL volumetric flasks, transfer 5.0, 7.5, 10.0, 12.5, and 15.0 mL of this solution. Dilute the contents of each flask with 0.125 N hydrochloric acid to volume, and mix to obtain the solutions having known concentrations of about 1.0, 1.5, 2.0, 2.5, and 3.0 µg of magnesium per mL.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with 0.125 N hydrochloric acid to obtain a solution having a concentration of about 2.5 µg of magnesium per mL.
Procedure—
Concomitantly determine the absorbances of the
Standard preparations and the
Assay preparation at the magnesium emission line at 285.2 nm with an atomic absorption spectrophotometer (see
Spectrophotometry and Light-Scattering 851) equipped with a magnesium hollow-cathode lamp and an air–acetylene flame, using 0.125 N hydrochloric acid as the blank. Plot the absorbances of the
Standard preparations versus concentration, in µg per mL, of magnesium, and draw the straight line best fitting the five plotted points. From the graph so obtained, determine the concentration,
C, in µg per mL, of magnesium in the
Assay preparation. Calculate the quantity, in mg, of magnesium (Mg) in each mL of the Oral Solution taken by the formula:
C(L/D),
in which
L is the labeled quantity, in mg per mL, of magnesium in the Oral Solution taken; and
D is the concentration, in µg per mL, of magnesium in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution.
Assay for manganese—
Manganese standard stock solution—
Transfer 1.0 g of manganese, accurately weighed, to a 1000-mL volumetric flask, dissolve in 20 mL of nitric acid, dilute with 6 N hydrochloric acid to volume, and mix.
Standard preparations—
Quantitatively dilute 10.0 mL of Manganese standard stock solution with water to 200.0 mL to obtain a standard solution having a known concentration of about 50 µg of manganese per mL. To separate 100-mL volumetric flasks, transfer 1.0, 1.5, 2.0, 3.0, and 4.0 mL of this solution, dilute the contents of each flask with 0.125 N hydrochloric acid to volume, and mix to obtain solutions having known concentrations of about 0.5, 0.75, 1.0, 1.5, and 2.0 µg of manganese per mL.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with 0.125 N hydrochloric acid to obtain a solution having a concentration of about 1.5 µg of manganese per mL.
Procedure—
Concomitantly determine the absorbances of the
Standard preparations and the
Assay preparation at the manganese emission line at 279.5 nm with an atomic absorption spectrophotometer (see
Spectrophotometry and Light-Scattering 851) equipped with a manganese hollow-cathode lamp and an air–acetylene flame, using 0.125 N hydrochloric acid as the blank. Plot the absorbances of the
Standard preparations versus concentration, in µg per mL, of manganese, and draw the straight line best fitting the five plotted points. From the graph so obtained, determine the concentration,
C, in µg per mL, of manganese in the
Assay preparation. Calculate the quantity, in mg, of manganese (Mn) in each mL of the Oral Solution taken by the formula:
C(L/D),
in which
L is the labeled quantity, in mg per mL, of manganese in the Oral Solution taken; and
D is the concentration, in µg per mL, of manganese in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution.
Assay for molybdenum—
Molybdenum standard stock solution—
Transfer 1.0 g of molybdenum wire, accurately weighed, to a 1000-mL volumetric flask, and dissolve in 50 mL of nitric acid, warming if necessary. Dilute with water to volume, and mix.
Standard preparations—
Quantitatively dilute 10.0 mL of Molybdenum standard stock solution with water to obtain a standard solution having a known concentration of about 100 µg of molybdenum per mL. To separate 100-mL volumetric flasks, transfer 0.5, 1.0, 1.5, 2.0, and 3.0 mL of this solution. Dilute the contents of each flask with 0.125 N hydrochloric acid to volume, and mix to obtain the solutions having known concentrations of about 0.5, 1.0, 1.5, 2.0, and 3.0 µg of molybdenum per mL.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with 0.125 N hydrochloric acid to obtain a solution having a concentration of about 1 µg of molybdenum per mL.
Procedure—
Concomitantly determine the absorbances of the
Standard preparations and the
Assay preparation at the molybdenum emission line at 313 nm with an atomic absorption spectrophotometer (see
Spectrophotometry and Light-Scattering 851) equipped with a molybdenum hollow-cathode lamp and a nitrous oxide–acetylene flame, using 0.125 N hydrochloric acid as the blank. Plot the absorbances of the
Standard preparations versus concentration, in µg per mL, of molybdenum, and draw the straight line best fitting the five plotted points. From the graph so obtained, determine the concentration,
C, in µg per mL, of molybdenum in the
Assay preparation. Calculate the quantity, in µg, of molybdenum (Mo) in each mL of the Oral Solution taken by the formula:
C(L/D),
in which
L is the labeled quantity, in µg per mL, of molybdenum in the Oral Solution taken; and
D is the concentration, in µg per mL, of molybdenum in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution.
Assay for zinc—
Zinc standard stock solution—
Transfer about 311 mg of zinc oxide, accurately weighed, to a 250-mL volumetric flask, and add 80 mL of 6 N hydrochloric acid, warming if necessary to dissolve. Cool, dilute with water to volume, and mix to obtain a solution having a known concentration of about 1000 µg of zinc per mL.
Standard preparations—
Dilute a volume of Zinc standard stock solution quantitatively, and stepwise if necessary, with water to obtain a standard solution having a known concentration of about 50 µg of zinc per mL. Transfer 1.0, 2.0, 3.0, 4.0, and 5.0 mL of this solution to separate 100-mL volumetric flasks. Dilute the contents of each flask with 0.125 N hydrochloric acid to volume, and mix to obtain the solutions having known concentrations of about 0.5, 1.0, 1.5, 2.0, and 2.5 µg of zinc per mL.
Assay preparation—
Dilute an accurately measured volume of Oral Solution quantitatively, and stepwise if necessary, with 0.125 N hydrochloric acid to obtain a solution having a concentration of about 1 µg of zinc per mL.
Procedure—
Concomitantly determine the absorbances of the
Standard preparations and the
Assay preparation at the zinc emission line at 213.8 nm with an atomic absorption spectrophotometer (see
Spectrophotometry and Light-Scattering 851) equipped with a zinc hollow-cathode lamp and an air–acetylene flame, using 0.125 N hydrochloric acid as the blank. Plot the absorbances of the
Standard preparations versus concentration,
C, in µg per mL, of zinc, and draw the straight line best fitting the five plotted points. From the graph so obtained, determine the concentration,
C, in µg per mL, of zinc in the
Assay preparation. Calculate the quantity, in mg, of zinc (Zn) in each mL of the Oral Solution taken by the formula:
C(L/D),
in which
L is the labeled quantity, in mg per mL, of zinc in the Oral Solution taken; and
D is the concentration, in µg per mL, of zinc in the
Assay preparation, on the basis of the labeled quantity and the extent of dilution.