ULTRAVIOLET, VISIBLE, INFRARED, ATOMIC ABSORPTION, FLUORESCENCE, TURBIDIMETRY, NEPHELOMETRY, AND RAMAN MEASUREMENT
Absorption spectrophotometry is the measurement of an interaction between electromagnetic radiation and the molecules, or atoms, of a chemical substance. Techniques frequently employed in pharmaceutical analysis include UV, visible, IR, and atomic absorption spectroscopy. Spectrophotometric measurement in the visible region was formerly referred to as colorimetry; however, it is more precise to use the term “colorimetry” only when considering human perception of color.
Fluorescence spectrophotometry is the measurement of the emission of light from a chemical substance while it is being exposed to UV, visible, or other electromagnetic radiation. In general, the light emitted by a fluorescent solution is of maximum intensity at a wavelength longer than that of the exciting radiation, usually by some 20 to 30 nm.
Light-Scattering involves measurement of the light scattered because of submicroscopic optical density inhomogeneities of solutions and is useful in the determination of weight-average molecular weights of polydisperse systems in the molecular weight range from 1000 to several hundred million. Two such techniques utilized in pharmaceutical analysis are turbidimetry and nephelometry.
Raman spectroscopy (inelastic light-scattering) is a light-scattering process in which the specimen under examination is irradiated with intense monochromatic light (usually laser light) and the light scattered from the specimen is analyzed for frequency shifts.
The wavelength range available for these measurements extends from the short wavelengths of the UV through the IR. For convenience of reference, this spectral range is roughly divided into the UV (190 to 380 nm), the visible (380 to 780 nm), the near-IR (780 to 3000 nm), and the IR (2.5 to 40 µm or 4000 to 250 cm
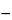 1
1).
COMPARATIVE UTILITY OF SPECTRAL RANGES
For many pharmaceutical substances, measurements can be made in the UV and visible regions of the spectrum with greater accuracy and sensitivity than in the near-IR and IR. When solutions are observed in 1-cm cells, concentrations of about 10 µg of the specimen per mL often will produce absorbances of 0.2 to 0.8 in the UV or the visible region. In the IR and near-IR, concentrations of 1 to 10 mg per mL and up to 100 mg per mL, respectively, may be needed to produce sufficient absorption; for these spectral ranges, cell lengths of from 0.01 mm to upwards of 3 mm are commonly used.
The UV and visible spectra of substances generally do not have a high degree of specificity. Nevertheless, they are highly suitable for quantitative assays, and for many substances they are useful as additional means of identification.
There has been increasing interest in the use of near-IR spectroscopy in pharmaceutical analysis, especially for rapid identification of large numbers of samples, and also for water determination.
The near-IR region is especially suitable for the determination of –OH and –NH groups, such as water in alcohol, –OH in the presence of amines, alcohols in hydrocarbons, and primary and secondary amines in the presence of tertiary amines.
The IR spectrum is unique for any given chemical compound with the exception of optical isomers, which have identical spectra. However, polymorphism may occasionally be responsible for a difference in the IR spectrum of a given compound in the solid state. Frequently, small differences in structure result in significant differences in the spectra. Because of the large number of maxima in an IR absorption spectrum, it is sometimes possible to quantitatively measure the individual components of a mixture of known qualitative composition without prior separation.
The Raman spectrum and the IR spectrum provide similar data, although the intensities of the spectra are governed by different molecular properties. Raman and IR spectroscopy exhibit different relative sensitivities for different functional groups, e.g., Raman spectroscopy is particularly sensitive to C–S and C–C multiple bonds, and some aromatic compounds are more easily identified by means of their Raman spectra. Water has a highly intense IR absorption spectrum, but a particularly weak Raman spectrum. Therefore, water has only limited IR “windows” that can be used to examine aqueous solutes, while its Raman spectrum is almost completely transparent and useful for solute identification. The two major limitations of Raman spectroscopy are that the minimum detectable concentration of specimen is typically 10
1 M to 10
2 M and that the impurities in many substances fluoresce and interfere with the detection of the Raman scattered signal.
Optical reflectance measurements provide spectral information similar to that obtained by transmission measurements. Since reflectance measurements probe only the surface composition of the specimen, difficulties associated with the optical thickness and the light-scattering properties of the substance are eliminated. Thus, reflectance measurements are frequently more simple to perform on intensely absorbing materials. A particularly common technique used for IR reflectance measurements is termed attenuated total reflectance (ATR), also known as multiple internal reflectance (MIR). In the ATR technique, the beam of the IR spectrometer is passed through an appropriate IR window material (e.g., KRS-5, a TlBr-TlI eutectic mixture), which is cut at such an angle that the IR beam enters the first (front) surface of the window, but is totally reflected when it impinges on the second (back) surface (i.e., the angle of incidence of the radiation upon the second surface of the window exceeds the critical angle for that material). By appropriate window construction, it is possible to have many internal reflections of the IR beam before it is transmitted out of the window. If a specimen is placed in close contact with the window along the sides that totally reflect the IR beam, the intensity of reflected radiation is reduced at each wavelength (frequency) that the specimen absorbs. Thus, the ATR technique provides a reflectance spectrum that has been increased in intensity, when compared to a simple reflectance measurement, by the number of times that the IR beam is reflected within the window. The ATR technique provides excellent sensitivity, but it yields poor reproducibility, and is not a reliable quantitative technique unless an internal standard is intimately mixed with each test specimen.
Fluorescence spectrophotometry is often more sensitive than absorption spectrophotometry. In absorption measurements, the specimen transmittance is compared to that of a blank; and at low concentrations, both solutions give high signals. Conversely, in fluorescence spectrophotometry, the solvent blank has low rather than high output, so that the background radiation that may interfere with determinations at low concentrations is much less. Whereas few compounds can be determined conveniently at concentrations below 10
5 M by light absorption, it is not unusual to employ concentrations of 10
7 M to 10
8 M in fluorescence spectrophotometry.
THEORY AND TERMS
The power of a radiant beam decreases in relation to the distance that it travels through an absorbing medium. It also decreases in relation to the concentration of absorbing molecules or ions encountered in that medium. These two factors determine the proportion of the total incident energy that emerge. The decrease in power of monochromatic radiation passing through a homogeneous absorbing medium is stated quantitatively by Beer's law, log10(1/T) = A = abc, in which the terms are as defined below.
Absorbance [Symbol: A]—
The logarithm, to the base 10, of the reciprocal of the transmittance (T). [NOTE—Descriptive terms used formerly include optical density, absorbancy, and extinction.]
Absorptivity [Symbol: a]—
The quotient of the absorbance (A) divided by the product of the concentration of the substance (c), expressed in g per L, and the absorption path length (b) in cm. [NOTE—It is not to be confused with absorbancy index; specific extinction; or extinction coefficient.]
Molar Absorptivity [Symbol: 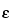 ]—
]—
The quotient of the absorbance
(A) divided by the product of the concentration, expressed in
moles per L, of the substance and the absorption path length in cm. It is also the product of the absorptivity
(a) and the molecular weight of the substance.
[NOTE—Terms formerly used include molar absorbancy index; molar extinction coefficient; and molar absorption coefficient.
]
For most systems used in absorption spectrophotometry, the absorptivity of a substance is a constant independent of the intensity of the incident radiation, the internal cell length, and the concentration, with the result that concentration may be determined photometrically.
Beer's law gives no indication of the effect of temperature, wavelength, or the type of solvent. For most analytical work the effects of normal variation in temperature are negligible.
Deviations from Beer's law may be caused by either chemical or instrumental variables. Apparent failure of Beer's law may result from a concentration change in solute molecules because of association between solute molecules or between solute and solvent molecules, or dissociation or ionization. Other deviations might be caused by instrumental effects such as polychromatic radiation, slit-width effects, or stray light.
Even at a fixed temperature in a given solvent, the absorptivity may not be truly constant. However, in the case of specimens having only one absorbing component, it is not necessary that the absorbing system conform to Beer's law for use in quantitative analysis. The concentration of an unknown may be found by comparison with an experimentally determined standard curve.
Although, in the strictest sense, Beer's law does not hold in atomic absorption spectrophotometry because of the lack of quantitative properties of the cell length and the concentration, the absorption processes taking place in the flame under conditions of reproducible aspiration do follow the Beer relationship in principle. Specifically, the negative log of the transmittance, or the absorbance, is directly proportional to the absorption coefficient, and, consequently, is proportional to the number of absorbing atoms. On this basis, calibration curves may be constructed to permit evaluation of unknown absorption values in terms of concentration of the element in solution.
Absorption Spectrum—
A graphic representation of absorbance, or any function of absorbance, plotted against wavelength or function of wavelength.
Transmittance [Symbol: T]—
The quotient of the radiant power transmitted by a specimen divided by the radiant power incident upon the specimen. [NOTE—Terms formerly used include transmittancy and transmission.]
Fluorescence Intensity [Symbol: I]—
An empirical expression of fluorescence activity, commonly given in terms of arbitrary units proportional to detector response. The
fluorescence emission spectrum is a graphical presentation of the spectral distribution of radiation emitted by an activated substance, showing intensity of emitted radiation as ordinate, and wavelength as abscissa. The
fluorescence excitation spectrum is a graphical presentation of the activation spectrum, showing intensity of radiation emitted by an activated substance as ordinate, and wavelength of the incident (activating) radiation as abscissa. As in absorption spectrophotometry, the important regions of the electromagnetic spectrum encompassed by the fluorescence of organic compounds are the UV, visible, and near-IR, i.e., the region from 250 to 800 nm. After a molecule has absorbed radiation, the energy can be lost as heat or released in the form of radiation of the same or longer wavelength as the absorbed radiation. Both absorption and emission of radiation are due to the transitions of electrons between different energy levels, or orbitals, of the molecule. There is a time delay between the absorption and emission of light; this interval, the duration of the excited state, has been measured to be about 10
9 second to 10
8 second for most organic fluorescent solutions. The short lifetime of fluorescence distinguishes this type of luminescence from phosphorescence, which is a long-lived afterglow having a lifetime of 10
3 second up to several minutes.
Turbidance [Symbol: S]—
The light-scattering effect of suspended particles. The amount of suspended matter may be measured by observation of either the transmitted light (turbidimetry) or the scattered light (nephelometry).
Turbidity [Symbol: 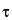 ]—
]—
In light-scattering measurements, the turbidity is the measure of the decrease in incident beam intensity per unit length of a given suspension.
Raman Scattering Activity—
The molecular property (in units of cm4 per g) governing the intensity of an observed Raman band for a randomly oriented specimen. The scattering activity is determined from the derivative of the molecular polarizability with respect to the molecular motion giving rise to the Raman shifted band. In general, the Raman band intensity is linearly proportional to the concentration of the analyte.
USE OF REFERENCE STANDARDS
With few exceptions, the Pharmacopeial spectrophotometric tests and assays call for comparison against a USP Reference Standard. This is to ensure measurement under conditions identical for the test specimen and the reference substance. These conditions include wavelength setting, slit-width adjustment, cell placement and correction, and transmittance levels. It should be noted that cells exhibiting identical transmittance at a given wavelength may differ considerably in transmittance at other wavelengths. Appropriate cell corrections should be established and used where required.
The expressions, “similar preparation” and “similar solution,” as used in tests and assays involving spectrophotometry, indicate that the reference specimen, generally a USP Reference Standard, is to be prepared and observed in a manner identical for all practical purposes to that used for the test specimen. Usually in making up the solution of the specified Reference Standard, a solution of about (i.e., within 10%) the desired concentration is prepared and the absorptivity is calculated on the basis of the exact amount weighed out; if a previously dried specimen of the Reference Standard has not been used, the absorptivity is calculated on the anhydrous basis.
The expressions, “concomitantly determine” and “concomitantly measured,” as used in tests and assays involving spectrophotometry, indicate that the absorbances of both the solution containing the test specimen and the solution containing the reference specimen, relative to the specified test blank, are to be measured in immediate succession.
APPARATUS
Many types of spectrophotometers are available. Fundamentally, most types, except those used for IR spectrophotometry, provide for passing essentially monochromatic radiant energy through a specimen in suitable form, and measuring the intensity of the fraction that is transmitted. Fourier transform IR spectrophotometers use an interferometric technique whereby polychromatic radiation passes through the analyte and onto a detector on an intensity and time basis. UV, visible, and dispersive IR spectrophotometers comprise an energy source, a dispersing device (e.g., a prism or grating), slits for selecting the wavelength band, a cell or holder for the test specimen, a detector of radiant energy, and associated amplifiers and measuring devices. In diode array spectrophotometers, the energy from the source is passed through the test specimen and then dispersed via a grating onto several hundred light-sensitive diodes, each of which in turn develops a signal proportional to the number of photons at its small wavelength interval; these signals then may be computed at rapid chosen intervals to represent a complete spectrum. Fourier transform IR systems utilize an interferometer instead of a dispersing device and a digital computer to process the spectral data. Some instruments are manually operated, whereas others are equipped for automatic and continuous recording. Instruments that are interfaced to a digital computer have the capabilities also of co-adding and storing spectra, performing spectral comparisons, and performing difference spectroscopy (accomplished with the use of a digital absorbance subtraction method).
Instruments are available for use in the visible; in the visible and UV; in the visible, UV, and near-IR; and in the IR regions of the spectrum. Choice of the type of spectrophotometric analysis and of the instrument to be used depends upon factors such as the composition and amount of available test specimen, the degree of accuracy, sensitivity, and selectivity desired, and the manner in which the specimen is handled.
The apparatus used in atomic absorption spectrophotometry has several unique features. For each element to be determined, a specific source that emits the spectral line to be absorbed should be selected. The source is usually a hollow-cathode lamp, the cathode of which is designed to emit the desired radiation when excited. Since the radiation to be absorbed by the test specimen element is usually of the same wavelength as that of its emission line, the element in the hollow-cathode lamp is the same as the element to be determined. The apparatus is equipped with an aspirator for introducing the test specimen into a flame, which is usually provided by air–acetylene, air–hydrogen, or, for refractory cases, nitrous oxide–acetylene. The flame, in effect, is a heated specimen chamber. A detector is used to read the signal from the chamber. Interfering radiation produced by the flame during combustion may be negated by the use of a chopped source lamp signal of a definite frequency. The detector should be tuned to this alternating current frequency so that the direct current signal arising from the flame is ignored. The detecting system, therefore, reads only the change in signal from the hollow-cathode source, which is directly proportional to the number of atoms to be determined in the test specimen. For Pharmacopeial purposes, apparatus that provides the readings directly in absorbance units is usually required. However, instruments providing readings in percent transmission, percent absorption, or concentration may be used if the calculation formulas provided in the individual monographs are revised as necessary to yield the required quantitative results. Percent absorption or percent transmittance may be converted to absorbance, A, by the following two equations:
A = 2
log
10 (100
% absorption)
A = 2
log
10 (% transmittance)
Depending upon the type of apparatus used, the readout device may be a meter, digital counter, recorder, or printer. Both single-beam and double-beam instruments are commercially available, and either type is suitable.
Measurement of fluorescence intensity can be made with a simple
filter fluorometer. Such an instrument consists of a radiation source, a primary filter, a specimen chamber, a secondary filter, and a fluorescence detection system. In most such fluorometers, the detector is placed on an axis at 90
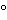
from that of the exciting beam. This right-angle geometry permits the exciting radiation to pass through the test specimen and not contaminate the output signal received by the fluorescence detector. However, the detector unavoidably receives some of the exciting radiation as a result of the inherent scattering properties of the solutions themselves, or if dust or other solids are present. Filters are used to eliminate this residual scatter. The primary filter selects short-wavelength radiation capable of exciting the test specimen, while the secondary filter is normally a sharp cut-off filter that allows the longer-wavelength fluorescence to be transmitted but blocks the scattered excitation.
Most fluorometers use photomultiplier tubes as detectors, many types of which are available, each having special characteristics with respect to spectral region of maximum sensitivity, gain, and electrical noise. The photocurrent is amplified and read out on a meter or recorder.
A spectrofluorometer differs from a filter fluorometer in that filters are replaced by monochromators, of either the prism or the grating type. For analytical purposes, the spectrofluorometer is superior to the filter fluorometer in wavelength selectivity, flexibility, and convenience, in the same way in which a spectrophotometer is superior to a filter photometer.
Many radiation sources are available. Mercury lamps are relatively stable and emit energy mainly at discrete wavelengths. Tungsten lamps provide an energy continuum in the visible region. The high-pressure xenon arc lamp is often used in spectrofluorometers because it is a high-intensity source that emits an energy continuum extending from the UV into the IR.
In spectrofluorometers, the monochromators are equipped with slits. A narrow slit provides high resolution and spectral purity, while a large slit sacrifices these for high sensitivity. Choice of slit size is determined by the separation between exciting and emitting wavelengths as well as the degree of sensitivity needed.
Specimen cells used in fluorescence measurements may be round tubes or rectangular cells similar to those used in absorption spectrophotometry, except that they are polished on all four vertical sides. A convenient test specimen size is 2 to 3 mL, but some instruments can be fitted with small cells holding 100 to 300 µL, or with a capillary holder requiring an even smaller amount of specimen.
Light-scattering instruments are available and consist in general of a mercury lamp, with filters for the strong green or blue lines, a shutter, a set of neutral filters with known transmittance, and a sensitive photomultiplier to be mounted on an arm that can be rotated around the solution cell and set at any angle from
135
to 0
to +135
by a dial outside the light-tight housing. Solution cells are of various shapes, such as square for measuring 90
scattering; semioctagonal for 45
, 90
, and 135
scattering; and cylindrical for scattering at all angles. Since the determination of molecular weight requires a precise measure of the difference in refractive index between the solution and solvent, [(
n n0)
/c], a second instrument, a differential refractometer, is needed to measure this small difference.
Raman spectrometers include the following major components: a source of intense monochromatic radiation (invariably a laser); optics to collect the light scattered by the test specimen; a (double) monochromator to disperse the scattered light and reject the intense incident frequency; and a suitable light-detection and amplification system. Raman measurement is simple in that most specimens are examined directly in melting-point capillaries. Because the laser source can be focused sharply, only a few microliters of the specimen is required.