Apparatus—
The apparatus for hydroxypropoxy group determinations is shown diagrammatically in
Fig. 1.
Fig. 1. Apparatus for hydroxypropoxy determination.
The boiling or reaction flask, consisting of a 125-mL conical-bottom boiling flask modified to provide a thermocouple (or thermometer) well and an inlet with a 1.0-mm capillary tip for nitrogen and water (see
Fig. 2), is fitted with a distillation head that leads to a condenser. The reaction flask is immersed in an oil bath equipped with an electric heater capable of heating the bath at the desired rate and maintaining the temperature at 155
. The distillate is collected in a flask.
[NOTE—The tube from the condenser to the flask must be below the surface of the liquid in the flask to ensure the capture of all of the acetic acid formed. See
Fig. 1.
]
Procedure—
Transfer about 65 mg of Hydroxypropyl Cellulose, previously dried at 105
for 1 hour and accurately weighed, into the reaction flask. Add 5 mL of water, and swirl gently for 5 minutes. Add 10 mL of chromium trioxide solution (30 g in 70 mL). Assemble the apparatus as shown in Figures 1 and 2, and immerse the reaction flask in the oil bath slightly above the level of the chromium trioxide solution. Start the condenser cooling water, and pass nitrogen gas through the flask at a rate of about 70 to 75 mL per minute. Raise the temperature of the oil bath to 155
during a 30-minute period, and maintain it at this temperature throughout the determination.
[NOTE—Too rapid an initial rise in temperature results in high blank determinations.
] Monitor the temperature of the reaction mixture in the reaction flask using a thermocouple or thermometer in a well, as shown in Figures 1 and 2. When a reaction mixture temperature of 102 ± 1
is reached, add water through the water inlet until the reaction mixture temperature drops to 97 ± 1
. Continue this 97
to 102
temperature cycle until 100 mL of distillate has been collected. Detach the condenser from the distillation head, and wash with water, collecting the washings in the flask containing the distillate. Titrate the solution with 0.02 N sodium hydroxide VS to a pH of 7.0 ± 0.1, using an expanded-scale pH meter equipped with glass and calomel electrodes. Record the volume,
V, of the 0.02 N sodium hydroxide used, then add 500 mg of sodium bicarbonate and 10 mL of 2 N sulfuric acid. After evolution of carbon dioxide has ceased, add 1 g of potassium iodide, insert the stopper in the flask, shake the mixture, and allow the solution to stand in the dark for 5 minutes. Titrate the liberated iodine with 0.02 N sodium thiosulfate VS to the sharp disappearance of the yellow iodine color, adding a few drops of
starch TS to confirm the endpoint. Record the volume,
Y, required. This titration,
Y mL, multiplied by the empirical factor,
K, appropriate to the particular apparatus and reagents in use (see calculation below), gives the acid equivalent not caused by acetic acid. The acetic acid equivalent is (
V 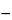 KY
KY) mL of 0.02 N sodium hydroxide.
in which VA is the volume, in mL, of 0.02 N sodium hydroxide required for titration of the sample; N1 is the normality of the 0.02 N sodium hydroxide; K is the empirical factor; YA is the volume, in mL, of 0.02 N sodium thiosulfate required for titration of the sample; N2 is the normality of the 0.02 N sodium thiosulfate; and W is the quantity, in g, of sample used. Each mL of 0.02 N sodium hydroxide is equivalent to 1.502 mg of hydroxypropoxy groups (–OCH2CHOHCH3).
;)
;)
;)