Histological characterization—
SOLUTION PREPARATIONS—
2.0 M Monobasic potassium phosphate—
Dissolve 13.61 g of anhydrous monobasic potassium phosphate in 50 mL of water.
2.0 M Dibasic potassium phosphate—
Dissolve 17.42 g of anhydrous dibasic potassium phosphate in 50 mL of water.
Phosphate-buffered saline solution (pH 7.1–7.5)—
Combine 3.6 mL of 2.0 M Monobasic potassium phosphate, 16.4 mL of 2.0 M Dibasic potassium phosphate, 8 g of sodium chloride, and 1 L of water. Mix thoroughly.
0.3% Acid alcohol—
To 100 mL of 70% alcohol, add 0.3 mL of hydrochloric acid, and mix.
Hematoxylin–alcohol solution—
Dissolve 2.5 g of hematoxylin in 25.0 mL of dehydrated alcohol, with heating.
Potassium alum solution—
Dissolve 50.0 g of potassium alum in 500 mL of water, with heating.
Hematoxylin staining solution—
Mix Hematoxylin–alcohol solution and Potassium alum solution, and heat to boiling as rapidly as possible with constant stirring. Do not heat for more than 1 minute. Slowly add 0.185 g of sodium iodate, and reheat to a simmer until the solution becomes a deep purple. Remove from the heat, and quickly cool. Filter daily before use.
Bluing agent—
Dissolve 200 mg of sodium bicarbonate and 40 mg of lithium carbonate in 63 mL of water and 37 mL of methanol, and mix.
Eosin solution—
Dissolve 1 g of eosin Y in 100 mL of alcohol. Filter daily before use.
TISSUE PREPARATION—
Remove three 2-cm diameter circular sections from every 30-cm
2 section of Graftskin (not less than 30% of the total unit area), using the appropriate size biopsy punch. Cut with a circular rocking motion to prevent crushing the tissue. Immerse the sections in 3.7% dimethoxymethane for 30 minutes, using a gentle rocking motion. Remove the sections, and lay on a cutting surface, dermal side (glossy side) down. Cut an approximately 3-mm-wide strip through the center of the specimen, using a new, single-edged razor blade. Place the strips in a histological microwave cassette, using suitable biopsy pads premoistened with
Phosphate-buffered saline solution (pH 7.1–7.5) to hold the strips in place. Insert the cassette into a histological microwave processing rack, place the rack inside a suitable microwave container, and add sufficient
Phosphate-buffered saline solution (pH 7.1–7.5) to completely cover the rack. Place the container in a microwave oven suitable for histological work,
2 and heat for 4 minutes at 55
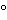
. Remove the
Phosphate-buffered saline solution (pH 7.1–7.5), and add enough dehydrated alcohol to completely cover the rack. Return the container to the microwave oven, and heat for 4 minutes at 67
. Remove the alcohol, and add enough dehydrated isopropyl alcohol to completely cover the rack. Return the container to the microwave oven, and heat for 4 minutes at 74
. Remove the isopropyl alcohol, and add enough suitable grade paraffin
3 that has been melted and held at 84
prior to use, to completely cover the rack. Return the container to the microwave oven, and heat for 7 minutes at 84
. Remove the histological microwave cassette from the container and rack while the paraffin is still melted, and disassemble, discarding the biopsy pads. Fill preheated embedding molds with molten paraffin
4 heated to 60
, and place on top of a preheated warming platform that is designed for histological work. Using forceps, remove the Graftskin specimens from the cassette, and place the specimens in individual molds. Orient the specimens in the molds to enable cutting of a cross-or longitudinal section. Cool the paraffin by sliding the mold down the platform to its cool side until the paraffin has solidified. Maintain the specimen orientation with forceps during cooling, removing the forceps when the paraffin becomes translucent. Slide the paraffin block onto a histological cold plate to rapidly cool the block. Trim the paraffin block with a new single-edged razor blade to form a rectangle or slight trapezoid to within 5 mm of the tissue mass, if necessary. Cool the block at 4
for 15 to 30 minutes, and clamp the paraffin block into the block holder of the microtome. Fill a histological tissue-flotation water bath with fresh water, add an appropriate amount of a suitable histological adhesive,
5 and heat to a temperature 5
lower than the melting point of the paraffin. Properly mount the paraffin block into a microtome, adjusting as necessary. Set the microtome to make 5-µm thick cuts with a blade angle of 5 ± 2
. Insert into the knife holder a sharp stainless steel microtome knife that has been properly honed or a new disposable microtome knife, and cut a ribbon that contains 6 to 10 sections of Graftskin. Pick up the ribbon with forceps, and stretch it across the tissue-flotation water bath. Separate 2 to 3 adjacent sections from the ribbon on the water bath. The selected sections should not be compressed, wrinkled, or scratched. Pick up the selected sections by dipping a microscope slide into the water bath under the floating sections, and gently lift the slide out of the water. Allow the mounted sections to air-dry completely, or dry the slide in a 60
oven for 1 hour. The microscope slide with affixed tissue is sequentially immersed in 3 changes of a suitable histological, aliphatic xylene substitute,
6 5 minutes per step, followed by two changes of dehydrated alcohol, 3 minutes per step. Sequentially immerse the slide in alcohol (for 3 minutes), running water rinse (3 minutes),
Hematoxylin staining solution (6 minutes), running water rinse (7 minutes),
0.3% Acid alcohol (6 seconds), running water rinse (5 minutes),
Bluing agent (1 second), running water rinse (5 minutes),
Eosin solution (2 minutes), 2 changes of alcohol (3 minutes each step), 4 changes of dehydrated alcohol (3 minutes each step), and 4 changes of a suitable histological xylene substitute (3 minutes each step). Adjust the above immersion times as needed to suitably stain the tissue. Remove the slide from the last histological xylene substitute wash, and blot dry the back of the slide. Do not allow the tissue to dry. Affix a coverslip over the tissue, using a suitable coverslip mountant.
MICROSCOPIC SPECIFICATIONS—
A light microscope with 4×, 10×, 20×, and 40× objectives installed in a revolving nosepiece; a 10× widefield ocular with 10 to 19 mm per 100 microdisk reticle installed; and a 10× widefield ocular with grid reticle installed.
MICROSCOPIC AND MORPHOLOGICAL CHARACTERISTICS—
Score the 3 Graftskin sections for epidermal and dermal aspects, using the light microscope. Evaluate the slides from each of the sections taken. Average the aspect values for each section (
n = 3) to determine the overall aspect score for the Graftskin unit. When examined microscopically, Graftskin shows a bilayered construct resembling the epidermal and dermal layers of human skin. Using
USP Graftskin Reference Photomicrographs of passing and failing articles for comparison, Graftskin meets the requirements for epidermal aspects, including epidermal coverage, epidermal development, and keratinocyte aspect, and meets the requirements for dermal aspects, including dermal matrix thickness, fibroblast density, and matrix aspect, as described below.
Epidermal aspects (see USP Graftskin Reference Photomicrograph 1 for an example of a passing unit)—
Epidermal coverage—
Ninety-five percent or more of the dermal matrix present on the slide is covered with epidermal keratinocytes.
Epidermal development—
Seventy percent or more of the Graftskin epithelium is composed of 3 distinct cell layers (see USP Graftskin Reference Photomicrograph 2 for an example of a failing unit). The basal cell layer of the epithelium is at least 1 cell thick, consisting of keratinocytes with a cuboidal-columnar shape (see USP Graftskin Reference Photomicrograph 3 for an example of a failing unit). The suprabasal layer is composed of stratified cells and is at least 5 cells thick. Suprabasal cells closest to the basal layer are cuboidal in shape; cells become progressively stratified the closer they are to the uppermost, squamous cell layer. The squamous cell layer on the apical surface is cornified and at least 1 cell thick (see USP Graftskin Reference Photomicrograph 4 for an example of a failing unit). The uppermost cell layer of the epithelium is analogous to the stratum corneum of human skin and is composed of one or more rows of flat, scaly cells that are nonliving and keratinized (see USP Graftskin Reference Photomicrograph 5 for an example of a failing unit).
Keratinocyte aspect—
Ninety-five percent or more of the basal keratinocytes have basophilic cytoplasm that neither has distinct vacuoles nor is necrotic (see USP Graftskin Reference Photomicrograph 6 for an example of a failing unit). Eighty percent or more of suprabasal cells (excluding those in the upper 20% of the cell layer closest to the squamous layer) have basophilic cytoplasm. Furthermore, these basophilic suprabasal cells do not have distinct vacuoles and are neither necrotic nor keratinized (see USP Graftskin Reference Photomicrographs 7 and 8 for examples of failing units).
Dermal aspects (see USP Graftskin Reference Photomicrograph 1 for an example of a passing unit)—
Five randomly selected fields per slide will be evaluated for dermal matrix thickness and fibroblast density. The 5 fields will be averaged to obtain the final value for each section.
Dermal matrix thickness—
The Graftskin dermal layer is not less than 40 µm thick and is composed of several rows of flat dermal cells.
Fibroblast density—
The dermal matrix contains an average of at least 4 nonpyknotic nuclei present per microscopic field (field = 20 grid squares of reticle when using the 10× widefield ocular and 40× objective).
Matrix aspect—
At least 95% of the dermal matrix collagen stains uniformly with no large holes or inclusions present (see USP Graftskin Reference Photomicrographs 9 and 10 for examples of failing units).
Gene expression profile—
RNA extraction solution—
Use an aqueous phenol and guanidine isothiocyanate solution suitable for RNA extraction.
7
DEPC-treated water—
Add 0.2 mL of diethylpyrocarbonate (DEPC) to 100 mL of sterile Purified Water, shake vigorously, and allow to stand for at least 12 hours. Autoclave the resulting solution for 15 minutes, using the liquid cycle, to inactivate residual DEPC. Prepare fresh as needed.
5X Reaction buffer—
Prepare a solution of potassium chloride, magnesium chloride, and tris(hydroxymethyl)aminomethane hydrochloride having concentrations of 375 mM, 15 mM, and 250 mM, respectively. Adjust to a pH of 8.3.
10X Reaction buffer—
Prepare a solution of potassium chloride and tris(hydroxymethyl)aminomethane hydrochloride having concentrations of 500 mM and 100 mM, respectively. Adjust to a pH of 8.3.
Oligo-deoxythymidine solution—
Prepare a 20-mM oligo-deoxythymidine (primer length: 18) solution, using a suitable buffer.
8
dNTP solution I—
Using a suitable buffer,
8 prepare a solution of deoxyadenosine triphosphate, deoxyguanosine triphosphate, deoxycytidine triphosphate, and deoxythymidine triphosphate in which the concentration of each component is 10 mM.
dNTP solution II—
Prepare a solution, in water, of deoxyadenosine triphosphate, deoxyguanosine triphosphate, deoxycytidine triphosphate, and deoxythymidine triphosphate, in which the concentration of each component is 10 mM.
Ribonuclease inhibitor solution—
Prepare a solution containing 40 units of ribonuclease inhibitor per mL of a suitable buffer.
8
Reverse transcriptase solution—
Prepare a solution containing 200 units of reverse transcriptase per µL of a solution of sodium chloride, edetate disodium, dithiothreitol, nonylphenol polyoxyethylene ether, glycerin, and tris(hydroxymethyl)aminomethane hydrochloride having concentrations of 0.1 M, 0.1 M, 1.0 M, 0.01%, 50%, and 200 mM, respectively. Adjust to a pH of 7.5.
DNA primer pairs—
Prepare individual 20-µM solutions of the following DNA primer pairs,
9 using deoxyribonuclease- and ribonuclease-free water.
DNA polymerase solution—
Prepare a solution containing 5 units of deoxyribonucleic acid polymerase per mL of a solution of potassium chloride, edetate disodium, dithiothreitol, polyoxyethylene (20) sorbitan monolaurate, nonylphenol polyoxyethylene ether, glycerin, and tris(hydroxymethyl)aminomethane hydrochloride, having concentrations of 100 mM, 0.1 mM, 1 mM, 0.5%, 0.5%, 50%, and 20 mM, respectively. Adjust to a pH of 8.0.
RNA extraction procedure—
Remove three 2-cm diameter circular sections from every 30 cm
2 of Graftskin (not less than 30% of the total unit area), using the appropriate size biopsy punch. Transfer each piece of tissue to individual polypropylene microcentrifuge tubes. Add 1.0 mL of
RNA extraction solution to each tube, homogenize by repetitive pipetting, and incubate the samples for 5 minutes at room temperature. To each tube add 0.2 mL of chloroform, mix on a vortex mixer, and centrifuge at 12,000
g for 15 minutes at 2
to 8
. Transfer the upper, aqueous phase to a second tube, add 0.5 mL of isopropanol, and incubate for 30 minutes to overnight at –20
. Centrifuge at 12,000
g for 15 minutes, discard the supernatants by aspiration, and add 75% alcohol to each pellet. Mix the sample on a vortex mixer, centrifuge at 12,000
g for 2 minutes, and discard the supernatants by aspiration without disturbing the RNA pellets. Recentrifuge at 12,000
g for 2 minutes, and remove the remaining supernatants with a small-volume (20 µL or smaller capacity) micropipet. Air-dry the pellets for 5 minutes at room temperature by keeping the microcentrifuge cap off, and resuspend each pellet in 50 µL of
DEPC-treated water. Bring absorbance into linear range by diluting 5 µL of each suspension with 195 µL of
DEPC-treated water. Transfer the samples to suitable quartz microplates or cuvettes and determine the absorbance of the RNA solution at wavelengths of 260 and 280 nm, using a spectrophotometer and
DEPC-treated water as the blank. The ratio of the absorbance at 260 versus 280 nm should be greater than or equal to 1.65. If this ratio is less than 1.65, mix the resuspended pellet by repetitive pipetting, and repeat the dilution step and absorbance measurement. If this fails to raise the absorbance ratio, repeat the RNA extraction for that sample by adding 1 mL of
RNA extraction solution, and proceed as above, beginning with “incubate the sample for 5 minutes at room temperature”. Determine the concentration of RNA, in µg per mL, using the following equation:
40AD,
in which
A is the absorbance at 260 nm, and
D is the dilution factor. Adjust the volume of the RNA solutions with additional
DEPC-treated water to bring the concentration of RNA to about 80 µg per mL. If the absorbance at 260 nm is less than 0.05, discard the sample, and repeat the RNA extraction on a fresh sample.
Synthesis of cDNA—
To separate, individual thin-walled polymerase chain reaction (PCR) tubes add 12.5 µL of the RNA solution from samples 1, 2, and 3 (3 reaction tubes total). Add 1 µL of
Oligo-deoxythymidine solution to each tube, and incubate at 72
for 2 minutes to anneal the oligo-deoxythymidine to the mRNA. Place the tubes in an ice bath, and to each tube add 4 µL of
5X Reaction buffer, 1 µL of
dNTP solution I, 0.5 µL of
Ribonuclease inhibitor solution, and 1 µL of
Reverse transcriptase solution. Incubate at 42
for 1 hour to synthesize cDNA, and then incubate at 94
for 5 minutes to inactivate the reverse transcriptase. To each tube add 80 µL of
DEPC-treated water, and mix.
Polymerase chain reaction amplification of cDNA—
For each of the five
DNA primer pairs, label five individual centrifuge tubes (five tubes total). Add the following to each centrifuge tube:
DEPC-treated water, 135.8 µL;
dNTP solution II, 10.5 µL;
10X Reaction buffer, 21 µL; the appropriate 5
¢ primer, 3.5 µL; the appropriate 3
¢ primer, 3.5 µL; and 25 mM magnesium chloride, 12.6 µL. Close, mix on a vortex mixer, and pulse spin in a microcentrifuge. Add 2.1 µL of
DNA polymerase solution to each centrifuge tube, and mix by repetitive pipetting. For each primer pair, transfer 27 µL of the resulting solution to five thin-walled PCR tubes. There should be a total of 25 PCR tubes. Add the following to the PCR tubes of each primer set:
| PCR tube number |
| 1 |
3 µL Graftskin sample 1 cDNA |
| 2 |
3 µL Graftskin sample 2 cDNA |
| 3 |
3 µL Graftskin sample 3 cDNA |
| 4 |
3 µL cDNA positive control10 |
| 5 Negative control |
3 µL DEPC-treated water |
Repeat for the remaining primer pairs. The positive control contains authentic cDNA of
Transforming growth factor 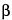 , Interleukin-1
, Interleukin-1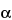 , Interleukin-4, Platelet-derived growth factor A,
, Interleukin-4, Platelet-derived growth factor A, and
Glyceraldehyde-3-phosphate dehydrogenase, as appropriate for each primer set. Pulse spin the PCR tubes in a microcentrifuge to mix, and place the tubes in a single PCR thermal cycler. Cycling conditions are as follows.
Terminate the PCR amplification by heating each tube to 72
for 7 minutes.
ELECTROPHORESIS IDENTIFICATION—
Tris-boric acid buffer—
Prepare a solution containing 89 mM of tris(hydroxymethyl) aminomethane, 89 mM of boric acid, and 2 mM of edetate disodium per L.
6X Loading buffer—
Prepare a solution containing 15% of a branched polymeric sucrose (400 kDa), 0.25% bromophenol blue, and 0.25% xylene cyanole FF.
Ethidium bromide solution—
Prepare a solution of ethidium bromide in Tris-boric acid buffer having a concentration of 10 mg per mL.
Agarose gel—
Prepare a horizontal 2% agarose
11 gel in
Tris-boric acid buffer. Once the gel is set, remove the comb, and place the gel into the electrophoresis chamber with the comb end of the gel situated closest to the cathode terminal. Fill the electrophoresis chamber with
Tris-boric acid buffer until the buffer reaches 3 to 5 mm over the surface of the gel.
100-bp DNA ladder markers—
Prepare a solution containing 10 DNA fragments covering the range of 100 to 1000 base pairs (bp) in 100-bp increments, with a total DNA content of approximately 100 ng per µL (15–20 ng of DNA per band) in an appropriate buffer.
12
Procedure—
Dilute the 25 PCR samples prepared in the
Polymerase chain reaction amplification of cDNA with
6X Loading buffer so that the final concentration of the buffer is one-sixth of its original concentration. Load 5 µL of the
100-bp DNA ladder markers in the first lane of the agarose gel. Load 10 µL of each PCR sample into each gel well, and attach the cathode to the terminal close to the loaded wells. Attach the anode to the terminal farthest from the loaded wells, and apply 120 V to the gel. Run the gel until the bromophenol blue is about two-thirds the length of the gel. Remove the gel from the electrophoresis apparatus, and place it in a tray containing enough
Ethidium bromide solution to cover the gel. Slowly agitate the gel on a shaker table for 30 minutes. Completely remove the
Ethidium bromide solution from the tray, add an equal amount of
Tris-boric acid buffer, and slowly agitate the gel on a shaker table for 60 minutes. Place the gel on a 312-nm UV light source, photograph the gel, and inspect the image for bands that have migrated from each individual well. If a band appears, it is verified for size in base pairs by comparing it to the lane for the 100-bp DNA ladder marker. If a band appears and it is of the appropriate size, it is considered positive. The analysis is considered valid if the positive controls show the appropriately sized cDNA–PCR products, no PCR product bands appear in the negative controls, and all bands are observed to be visually discrete. The lanes of the agarose gel that correspond to Graftskin show cDNA bands for
Interleukin-1 (expected PCR product band size of 491 base pairs, limit of detection not less than 9.6 ×10
–21 moles);
Platelet-derived growth factor (expected PCR product band size of 304 base pairs, limit of detection not less than 1.5 ×10
–20 moles);
Transforming growth factor-1 (expected PCR product band size of 161 base pairs, limit of detection not less than 1.5 × 10
–20) moles; and
Glyceraldehyde-3-phosphate dehydrogenase (expected PCR product band size of 983 base pairs); but not
Interleukin-4 (expected PCR product band size of 344 base pairs, limit of detection not less than 1.5 × 10
–22 moles). If one of the replicates tested yields results discordant with the other two replicates, repeat the assay, and accept only if all 3 replicates are concordant.
Barrier integrity assessment—
Ham's F-12 tissue culture medium—
Prepare a solution that contains the following:
| Component |
mg per mL |
| L-Alanine |
8.91 |
| L-Arginine hydrochloride |
210.7 |
| L-Asparagine monohydrate |
15.01 |
| L-Aspartic acid |
13.30 |
L-Cysteine hydrochloride
 monohydrate monohydrate |
35.12 |
| L-Glutamic acid |
14.70 |
| L-Glutamine |
146.2 |
| Aminoacetic acid |
7.51 |
L-Histidine hydrochloride
monohydrate |
20.96 |
| L-Isoleucine |
3.94 |
| L-Leucine |
13.12 |
| L-Lysine hydrochloride |
36.54 |
| L-Methionine |
4.48 |
| L-Phenylalanine |
4.96 |
| L-Proline |
34.53 |
| L-Serine |
10.51 |
| L-Threonine |
11.91 |
| L-Tryptophan |
2.04 |
| L-Tyrosine disodium |
6.71 |
| L-Valine |
11.71 |
| Calcium chloride |
44.00 |
| Cupric sulfate, pentahydrate |
0.0025 |
| Ferric sulfate, heptahydrate |
0.834 |
| Potassium chloride |
223.7 |
| Magnesium chloride |
57.22 |
| Sodium chloride |
7599.0 |
| Sodium phosphate, dibasic |
142.0 |
| Zinc sulfate, heptahydrate |
0.863 |
| D-Biotin |
0.0073 |
| D-Calcium pantothenate |
0.238 |
| Choline chloride |
13.96 |
| Folic acid |
1.30 |
| Hypoxanthine |
4.04 |
| Inositol |
18.02 |
| Niacinamide |
0.0366 |
| Pyridoxine hydrochloride |
0.0617 |
| Riboflavin |
0.0376 |
| Thiamine hydrochloride |
0.337 |
| Thymidine |
0.727 |
| Cyanocobalamin |
1.36 |
| -Lipoic acid |
0.206 |
| Linoleic acid |
0.0841 |
| Dextrose |
1801.6 |
| Phenol red, sodium |
1.30 |
| Sodium pyruvate |
110.0 |
| Putrescine dihydrochloride |
0.161 |
| Sodium bicarbonate |
1176.0 |
Percutaneous absorption apparatus—
Prepare the apparatus as described below.
13
Six-well cell culture plate—
The dimensions are inner diameter, about 35 mm; depth, about 18 mm.
Cell culture well insert—
Each well is a plastic cylinder with inner length, about 15 mm; inner diameter, about 24 mm; outer diameter, about 27 mm, with a flanged end extending about 4 mm from the outer diameter. The inner diameter opposite the flanged end is covered by a taut polycarbonate membrane having a porosity of 3 µm. The flange should allow the Cell culture well insert to be suspended in the well of a Six-well cell culture plate, leaving a 3-mm space between the bottom of the Cell culture well insert and the inner bottom surface of the Six-well cell culture plate.
Percutaneous absorption insert—
Use a polytetrafluoroethylene cylinder having the following dimensions: length, about 20 mm; inner diameter, about 20 mm; outer diameter, about 23 mm with a flanged end extending about 3 mm from the outer diameter. Ten mm from the flanged end of the cylinder, the inner diameter begins to funnel so that the inner diameter at about 10 mm from the flanged end is about 20 mm, and the inner diameter at about 15 mm from the flanged end is about 8 mm. From about 15 mm to about 20 mm from the flanged end, the inner diameter remains at 8 mm. The outer diameter of the cylinder remains constant at about 23 mm. The flanged end is considered to be the top of the component.
Silicon grease—
Use high-vacuum silicon grease suitable for glass.
14
Procedure—
Fill each well of the
Six-well cell culture plate with 1.5 mL of
Ham's F-12 tissue culture medium. Remove two 2-cm circular sections from every 30 cm
2 of Graftskin (not less than 20% of the total unit area), using the appropriate size biopsy punch. Transfer each excised section to a separate
Cell culture well insert, dermal side down on the polycarbonate membrane. Using forceps, gently smooth out the section to remove any wrinkles. Apply a narrow ring of
Silicon grease to the underside of the
Percutaneous absorption insert, and place the insert into the
Cell culture well insert, grease side down, onto the epidermal surface of the Graftskin biopsy, with slight pressure to form a tight seal. Do not allow any grease to enter the 8-mm diameter exposed area of the Graftskin surface. Place the
Cell culture well insert containing the
Percutaneous absorption insert into one of the wells of the
Six-well cell culture plate containing 1.5 mL of
Ham's F-12 tissue culture medium. Apply 1.0 mL of
Tritiated water to the exposed surface of the Graftskin unit in the
Percutaneous absorption insert, and incubate at ambient temperature for 6 hours. At the end of each hour, transfer the
Cell culture well insert containing the
Percutaneous absorption insert to a new well within the
Six-well cell culture plate containing 1.5 mL of fresh
Ham's F-12 tissue culture medium. After the 6-hour incubation, remove the
Cell culture well insert. Remove a 0.5-mL aliquot of
Ham's F-12 tissue culture medium from each well of the
Six-well cell culture plate, and transfer into individual scintillation vials. Dispense 0.5 mL of
Tritiated water to a separate scintillation vial as a control; to each scintillation vial add 4.5 mL of a suitable scintillation cocktail,
15 and gently mix. Place the scintillation vials into a liquid scintillation counter, and count the emissions in the tritium spectrum for 60 seconds. Average the counts for each of the six time points (punch average) and duplicate sections (unit average). Determine the percent penetration per hour by the formula:
150(CS/CC),
in which
CS are the counts per minute of the 0.5-mL aliquot of the
Ham's F-12 tissue culture medium taken at the end of the incubation period; and
CC are the counts per minute in the 0.5-mL aliquot of
Tritiated water. Not more than 1.97% penetration is found.
Metabolic activity assessment—
Dulbecco's modified Eagle's tissue culture medium—
Prepare a solution that contains the following components.
| Component |
mg per L |
| Calcium chloride |
264.9 |
| Ferric nitrate, nonahydrate |
0.10 |
| Potassium chloride |
400.0 |
| Magnesium sulfate, heptahydrate |
200.0 |
| Sodium chloride |
6,400.0 |
| Sodium bicarbonate |
3,700.0 |
Sodium phosphate, monobasic
(monohydrate) |
125.0 |
| Dextrose |
4,500.0 |
| Phenol red |
15.0 |
| Sodium pyruvate |
110.0 |
| L-Arginine hydrochloride |
84.0 |
| L-Cystine |
48.0 |
| Aminoacetic acid |
30.0 |
| L-Histidine hydrochloride monohydrate |
42.0 |
| L-Isoleucine |
104.8 |
| L-Leucine |
104.8 |
| L-Lysine hydrochloride |
146.2 |
| L-Methionine |
30.0 |
| L-Phenylalanine |
66.0 |
| L-Serine |
42.0 |
| L-Threonine |
95.2 |
| L-Tryptophan |
16.0 |
| L-Tyrosine |
72.0 |
| L-Valine |
93.6 |
| D-Calcium pantothenate |
4.0 |
| Choline chloride |
4.0 |
| Folic acid |
4.0 |
| Inositol |
7.0 |
| Nicotinamide |
4.0 |
| Pyridoxine hydrochloride |
4.0 |
| Riboflavin |
0.40 |
| Thiamine hydrochloride |
4.0 |
MTT solution—
Dissolve 0.33 g of (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide in 1 L of Dulbecco's modified Eagle's tissue culture medium, with constant stirring. Pass the solution through a suitable size filter having a 0.2-µm porosity.
0.04 N Acidified isopropyl alcohol—
Add 3.45 mL of hydrochloric acid to 1 L of isopropyl alcohol, and mix thoroughly. Store at room temperature no longer than 6 months.
Procedure—
Immerse the Graftskin in separate 40.0-mL portions of
MTT solution, making sure that about 20 mL of
MTT solution is under the test article, and 20 mL of
MTT solution is on the surface. Take care not to produce any bubbles. Incubate for 3 hours at 37º, in an environment of air enriched with 10% carbon dioxide. After incubation, remove from the 37º, 10% carbon dioxide-enriched air environment. Transfer the Graftskin to a suitable cutting surface, and, using an appropriate biopsy punch, remove three 8-mm diameter circular sections from every 30 cm
2 of Graftskin (5% of unit area). Transfer each punch to individual snap-top test tubes. Add 0.9 mL of
0.04 N Acidified isopropyl alcohol to each tube, making sure that the tissue is completely submerged. If not submerged, use forceps to place the sample into the
0.04 N Acidified isopropyl alcohol. Cap each tube tightly, place on an orbital shaker, and shake for 1 hour at a moderate setting. After 1 hour, remove the tubes from the orbital shaker, and mix each tube on a vortex mixer. Inspect the tubes to make sure that the tissue samples continue to be submerged. If not, use forceps or another device to resubmerge the tissues. Return the tubes to the orbital shaker, and continue to shake for an additional 1 hour. Remove the tubes from the orbital shaker, mix the tubes on a vortex mixer, and transfer a 0.2-mL aliquot to a suitable 96-well flat-bottom plate. Read the absorbance of each sample at 570 nm, using 0.2 mL of
0.04 N Acidified isopropyl alcohol as the blank. The average absorbance value is
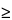
0.237.