Apparatus—
A specification for a definite size or type of container or apparatus in a test or assay is given solely as a recommendation. Where volumetric flasks or other exact measuring, weighing, or sorting devices are specified, this or other equipment of at least equivalent accuracy shall be employed. (See also
Thermometers  21
21
,
Volumetric Apparatus 31, and
Weights and Balances 41.) Where low-actinic or light-resistant containers are specified, clear containers that have been rendered opaque by application of a suitable coating or wrapping may be used.
Where an instrument for physical measurement, such as a spectrophotometer, is specified in a test or assay by its distinctive name, another instrument of equivalent or greater sensitivity and accuracy may be used. In order to obtain solutions having concentrations that are adaptable to the working range of the instrument being used, solutions of proportionately higher or lower concentrations may be prepared according to the solvents and proportions thereof that are specified for the procedure.
Where a particular brand or source of a material, instrument, or piece of equipment, or the name and address of a manufacturer or distributor, is mentioned (ordinarily in a footnote), this identification is furnished solely for informational purposes as a matter of convenience, without implication of approval, endorsement, or certification. Items capable of equal or better performance may be used if these characteristics have been validated.
Where the use of a centrifuge is indicated, unless otherwise specified, the directions are predicated upon the use of apparatus having an effective radius of about 20 cm (8 inches) and driven at a speed sufficient to clarify the supernatant layer within 15 minutes.
Unless otherwise specified, for chromatographic tubes and columns the diameter specified refers to internal diameter (ID); for other types of tubes and tubing the diameter specified refers to outside diameter (OD).
Steam Bath—
Where the use of a steam bath is directed, exposure to actively flowing steam or to another form of regulated heat, corresponding in temperature to that of flowing steam, may be used.
Water Bath—
Where the use of a water bath is directed without qualification with respect to temperature, a bath of vigorously boiling water is intended.
Procedures—
Assay and test procedures are provided for determining compliance with the Pharmacopeial standards of identity, strength, quality, and purity.
In performing the assay or test procedures in this Pharmacopeia, it is expected that safe laboratory practices will be followed. This includes the use of precautionary measures, protective equipment, and work practices consistent with the chemicals and procedures used. Prior to undertaking any assay or procedure described in this Pharmacopeia, the individual should be aware of the hazards associated with the chemicals and the procedures and means of protecting against them. This Pharmacopeia is not designed to describe such hazards or protective measures.
Every compendial article in commerce shall be so constituted that when examined in accordance with these assay and test procedures, it meets all the requirements in the monograph defining it. However, it is not to be inferred that application of every analytical procedure in the monograph to samples from every production batch is necessarily a prerequisite for ensuring compliance with Pharmacopeial standards before the batch is released for distribution. Data derived from manufacturing process validation studies and from in-process controls may provide greater assurance that a batch meets a particular monograph requirement than analytical data derived from an examination of finished units drawn from that batch. On the basis of such assurances, the analytical procedures in the monograph may be omitted by the manufacturer in judging compliance of the batch with the Pharmacopeial standards.
Automated procedures employing the same basic chemistry as those assay and test procedures given in the monograph are recognized as being equivalent in their suitability for determining compliance. Conversely, where an automated procedure is given in the monograph, manual procedures employing the same basic chemistry are recognized as being equivalent in their suitability for determining compliance. Compliance may be determined also by the use of alternative methods, chosen for advantages in accuracy, sensitivity, precision, selectivity, or adaptability to automation or computerized data reduction or in other special circumstances. Such alternative or automated procedures or methods shall be validated. However, Pharmacopeial standards and procedures are interrelated; therefore, where a difference appears or in the event of dispute, only the result obtained by the procedure given in this Pharmacopeia is conclusive.
In the performance of assay or test procedures, not fewer than the specified number of dosage units should be taken for analysis. Proportionately larger or smaller quantities than the specified weights and volumes of assay or test substances and Reference Standards may be taken, provided the measurement is made with at least equivalent accuracy and provided that any subsequent steps, such as dilutions, are adjusted accordingly to yield concentrations equivalent to those specified and are made in such manner as to provide at least equivalent accuracy. To minimize environmental impact or contact with hazardous materials, apparatus and chemicals specified in Pharmacopeial procedures also may be proportionally changed.
Where it is directed in an assay or a test that a certain quantity of substance or a counted number of dosage units is to be examined, the specified quantity or number is a minimal figure (the singlet determination) chosen only for convenience of analytical manipulation; it is not intended to restrict the total quantity of substance or number of units that may be subjected to the assay or test or that should be tested in accordance with good manufacturing practices.
Where it is directed in the assay of Tablets to “weigh and finely powder not fewer than” a given number, usually 20, of the Tablets, it is intended that a counted number of Tablets shall be weighed and reduced to a powder. The portion of the powdered tablets taken for assay is representative of the whole Tablets and is, in turn, weighed accurately. The result of the assay is then related to the amount of active ingredient per Tablet by multiplying this result by the average Tablet weight and dividing by the weight of the portion taken for the assay.
Similarly, where it is directed in the assay of Capsules to remove, as completely as possible, the contents of not fewer than a given number, usually 20, of the Capsules, it is intended that a counted number of Capsules should be carefully opened and the contents quantitatively removed, combined, mixed, and weighed accurately. The portion of mixed Capsules contents taken for the assay is representative of the contents of the Capsules and is, in turn, weighed accurately. The result of the assay is then related to the amount of active ingredient per Capsule by multiplying this result by the average weight of Capsule content and dividing by the weight of the portion taken for the assay.
Where the definition in a monograph states the tolerances as being “calculated on the dried (or anhydrous or ignited) basis,” the directions for drying or igniting the sample prior to assaying are generally omitted from the Assay procedure. Assay and test procedures may be performed on the undried or unignited substance and the results calculated on the dried, anhydrous, or ignited basis, provided a test for Loss on drying, or Water, or Loss on ignition, respectively, is given in the monograph. Results are calculated on an “as-is” basis unless otherwise specified in the monograph. Where the presence of moisture or other volatile material may interfere with the procedure, previous drying of the substance is specified in the individual monograph and is obligatory.
Throughout a monograph that includes a test for Loss on drying or Water, the expression “previously dried” without qualification signifies that the substance is to be dried as directed under Loss on drying or Water (gravimetric determination).
Unless otherwise directed in the test or assay in the individual monograph or in a general chapter, USP Reference Standards are to be dried before use, or used without prior drying, specifically in accordance with the instructions given in the chapter
USP Reference Standards 11, and on the label of the Reference Standard. Where the label instructions differ in detail from those in the chapter, the label text is determinative.
In stating the appropriate quantities to be taken for assays and tests, the use of the word “about” indicates a quantity within 10% of the specified weight or volume. However, the weight or volume taken is accurately determined, and the calculated result is based upon the exact amount taken. The same tolerance applies to specified dimensions.
Where the use of a pipet is directed for measuring a specimen or an aliquot in conducting a test or an assay, the pipet conforms to the standards set forth under
Volumetric Apparatus 31, and is to be used in such manner that the error does not exceed the limit stated for a pipet of its size. Where a pipet is specified, a suitable buret, conforming to the standards set forth under
Volumetric Apparatus 31, may be substituted. Where a “to contain” pipet is specified, a suitable volumetric flask may be substituted.
Expressions such as “25.0 mL” and “25.0 mg,” used with respect to volumetric or gravimetric measurements, indicate that the quantity is to be “accurately measured” or “accurately weighed” within the limits stated under
Volumetric Apparatus 31 or under
Weights and Balances 41.
The term “transfer” is used generally to specify a quantitative manipulation.
Blank Determination—
Where it is directed that “any necessary correction” be made by a blank determination, the determination is to be conducted using the same quantities of the same reagents treated in the same manner as the solution or mixture containing the portion of the substance under assay or test, but with the substance itself omitted.
Desiccator—
The expression “in a desiccator” specifies the use of a tightly closed container of suitable size and design that maintains an atmosphere of low moisture content by means of silica gel or other suitable desiccant.
A “vacuum desiccator” is one that maintains the low-moisture atmosphere at a reduced pressure of not more than 20 mm of mercury or at the pressure designated in the individual monograph.
Dilution—
Where it is directed that a solution be diluted “quantitatively and stepwise,” an accurately measured portion is to be diluted by adding water or other solvent, in the proportion indicated, in one or more steps. The choice of apparatus to be used should take into account the relatively larger errors generally associated with using small-volume volumetric apparatus (see
Volumetric Apparatus 31).
Drying to Constant Weight—
The specification “dried to constant weight” means that the drying shall be continued until two consecutive weighings do not differ by more than 0.50 mg per g of substance taken, the second weighing following an additional hour of drying.
Filtration—
Where it is directed to “filter,” without further qualification, the intent is that the liquid be passed through suitable filter paper or equivalent device until the filtrate is clear.
Identification Tests—
The Pharmacopeial tests headed Identification are provided as an aid in verifying the identity of articles as they are purported to be, such as those taken from labeled containers. Such tests, however specific, are not necessarily sufficient to establish proof of identity; but failure of an article taken from a labeled container to meet the requirements of a prescribed identification test indicates that the article may be mislabeled. Other tests and specifications in the monograph often contribute to establishing or confirming the identity of the article under examination.
Ignition to Constant Weight—
The specification “ignite to constant weight” means that the ignition shall be continued, at 800 ± 25
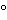
unless otherwise indicated, until two consecutive weighings do not differ by more than 0.50 mg per g of substance taken, the second weighing following an additional 15-minute ignition period.
Indicators—
Where the use of a test solution (“TS”) as an indicator is specified in a test or an assay, approximately 0.2 mL, or 3 drops, of the solution shall be added, unless otherwise directed.
Logarithms—
Logarithms used in the assays are to the base 10.
Microbial Strains—
Where a microbial strain is cited and identified by its ATCC catalog number, the specified strain shall be used directly or, if subcultured, shall be used not more than five passages removed from the original strain.
Negligible—
This term indicates a quantity not exceeding 0.50 mg.
Odor—
Terms such as “odorless,” “practically odorless,” “a faint characteristic odor,” or variations thereof, apply to examination, after exposure to the air for 15 minutes, either of a freshly opened package of the article (for packages containing not more than 25 g) or (for larger packages) of a portion of about 25 g of the article that has been removed from its package to an open evaporating dish of about 100-mL capacity. An odor designation is descriptive only and is not to be regarded as a standard of purity for a particular lot of an article.
Pressure Measurements—
The term “mm of mercury” used with respect to measurements of blood pressure, pressure within an apparatus, or atmospheric pressure refers to the use of a suitable manometer or barometer calibrated in terms of the pressure exerted by a column of mercury of the stated height.
Solutions—
Unless otherwise specified in the individual monograph, all solutions called for in tests and assays are prepared with
Purified Water.
An expression such as “(1 in 10)” means that 1 part by volume of a liquid is to be diluted with, or 1 part by weight of a solid is to be dissolved in, sufficient of the diluent or solvent to make the volume of the finished solution 10 parts by volume.
An expression such as “(20:5:2)” means that the respective numbers of parts, by volume, of the designated liquids are to be mixed, unless otherwise indicated.
The notation “VS” after a specified volumetric solution indicates that such solution is standardized in accordance with directions given in the individual monograph or under Volumetric Solutions in the section Reagents, Indicators, and Solutions, and is thus differentiated from solutions of approximate normality or molarity.
Where a standardized solution of a specific concentration is called for in a test or an assay, a solution of other normality or molarity may be used, provided allowance is made for the difference in concentration and provided the error of measurement is not increased thereby.
Specific Gravity—
Unless otherwise stated, the specific gravity basis is 25
/25
, i.e., the ratio of the weight of a substance in air at 25
to the weight of an equal volume of water at the same temperature.
Temperatures—
Unless otherwise specified, all temperatures in this Pharmacopeia are expressed in centigrade (Celsius) degrees, and all measurements are made at 25
. Where moderate heat is specified, any temperature not higher than 45
(113
F) is indicated. See
Storage Temperature under
Preservation, Packaging, Storage, and Labeling for other definitions.
Time Limit—
In the conduct of tests and assays, 5 minutes shall be allowed for the reaction to take place unless otherwise specified.
Vacuum—
The term “in vacuum” denotes exposure to a pressure of less than 20 mm of mercury unless otherwise indicated.
Where drying in vacuum over a desiccant is directed in the individual monograph, a vacuum desiccator or a vacuum drying pistol, or other suitable vacuum drying apparatus, is to be used.
Water—
Where water is called for in tests and assays,
Purified Water is to be used unless otherwise specified. For special kinds of water such as “carbon dioxide–free water,” see the introduction to the section
Reagents, Indicators, and Solutions. For
High-Purity Water see
Containers 661.
Water and Loss on Drying—
Where the water of hydration or adsorbed water of a Pharmacopeial article is determined by the titrimetric method, the test is generally given under the heading Water. Monograph limits expressed as a percentage are figured on a weight/weight basis unless otherwise specified. Where the determination is made by drying under specified conditions, the test is generally given under the heading Loss on drying. However, Loss on drying is most often given as the heading where the loss in weight is known to represent residual volatile constituents, including organic solvents as well as water.