General Procedure—
Plant samples are observed under the microscope by employing different mounting media, stains, or other solutions to assist in the correct identification of the test article. If a USP Authenticated Reference Material is available, prepare it with the same mounting media or reagent solutions used for the test article. Place one or two drops of water,
Glycerin–Alcohol Solution,
Chloral Hydrate Solution, or another reagent solution (see P
reparation and Use of Reagent Solutions,
Optical Devices,
and Mountants) in the center of a clean slide. Transfer a small plant tissue section or a portion of plant powder into the mountant or reagent solution, and cover with a clean coverslip. (For specific preparation techniques, see
Preparation of Temporary Mounts and Hand Sections,
Maceration, or
Preparation of Powdered Material, as appropriate.) To prevent the formation of air bubbles, the coverslip may be carefully placed at an appropriate angle with its edge making the first contact with the slide and then pressed until it covers the specimen. Using a piece of filter paper, remove excess fluid from the margin of the coverslip. Air bubbles can be removed by placing the slide in a vacuum desiccator. When using chloral hydrate, air bubbles can be removed by gently boiling the sample over a small flame such as that from an alcohol lamp. To replace the mountant or reagent solution, place drops of the new mountant or reagent solution on one edge of the coverslip. Place a strip of filter paper at the opposite edge of the coverslip to remove the old mountant or reagent solution and to cause the new mountant or reagent solution to be drawn over the powdered material or tissue. Plant oils can be also washed away from the tissue in this manner, when solvent hexane or acetone is washed through the slide followed by water and, if necessary,
Chloral Hydrate Solution. Do not use
Chloral Hydrate Solution immediately after treating the plant tissue with flammable solvents without thoroughly washing the tissue with water. This is to avoid setting fire to residual solvent when the microscope slide is later placed over a small flame to boil the tissue. Care must be taken when using reagent solutions that are volatile or corrosive to the microscope. To prevent drying of aqueous or chloral hydrate solutions during observation, add a small drop of glycerin to the slide. Observe the mounted sample under an optical microscope (see
Optical Microscopy  776
776
), and examine for histological features.
Preparation and Use of Reagent Solutions, Optical Devices, and Mountants—
The following reagents, optical devices, and mounting media are used to assist in the identification of cells, tissues, structural features, and ergastic substances in the tissue or powdered material (see
Tables 1 and
2).
Table 1. The Use of Reagent Solutions and Optical Devices
Table 2. Bleaching and Clarifying Agents and Mountants
| Use |
Mountants and Agents |
| Bleaching Agents |
Sodium Hypochlorite Solution |
| Clarifying Agents |
Chloral Hydrate Solution
Lactochloral Solution
Lactophenol Solution |
| Mountants |
Glycerin
Glycerin–Alcohol Solution
Glycerin–Gelatin Mixture
Water |
Alcoholic Picric Acid Solution—
Prepare a 1% solution of picric acid in alcohol. Picric acid is useful to stain cells having dense cytoplasm, such as aleurone cells in seeds. Place a small amount of powdered plant material in a test tube, and shake with about 1 mL of solvent hexane to remove plant oils, which would interfere with the reaction. Centrifuge, and discard solvent hexane. Soak the plant powder in Alcoholic Picric Acid Solution for about 30 minutes. Transfer a portion of the powder to a microscope slide, and observe under a microscope: cytoplasm and protein bodies turn bright yellow. [Caution—Picric acid is explosive when dry. Handle appropriately.
]
Blood–Gelatin Mixture—
Add 4.5 g of gelatin powder to 100 mL of a 0.9% sodium chloride solution, and allow to swell for 30 minutes. Heat the gel, while stirring, to about 80
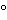
in a water bath. Cool to 40
, and add 6 mL of defibrinated bovine blood. Heat to 45
to 50
, and pour onto a microscope slide in a thin layer of about 1 mm while the slide is in a horizontal position. To prevent loss of blood-gelatin mixture from the sides, seal the microscope slide edge with a 1-cm wide adhesive tape to form a tray. After cooling and solidification, it is ready for use.
[NOTE—Store in a humid chamber for not more than 1 to 2 days at 3
to 4
.
] To test for saponins, place small clusters of the powdered plant material on the blood–gelatin layer, spacing them a few millimeters apart from each other, transfer to a humidifier for a few hours, and observe: saponin-containing particles will cause light-transparent zones to appear in the blood–gelatin.
Carmine Alum–Methyl Green Solution—
Boil 1.5 g of carmine for 30 minutes in a 15% solution of aluminum potassium sulfate. Cool, filter, and add 10 mL of a 0.75% methyl green solution while stirring. Add 1 to 2 drops to plant material: lignin and suberin turn green and cellulose turns red-violet.
Chloral Hydrate Solution—
Use chloral hydrate TS. When using the solution as a clarifying agent, add a few drops to the plant material, and boil briefly over a small flame. Chloral hydrate dissolves cellular contents and intercellular substances and allows cell walls and shapes to be easily observed. It can be used to assist in the identification of cork, fibers, vessels, calcium oxalate crystals (with the aid of crossed polarizers), trichomes, stomata, and pollen.
Crossed Polarizers—
This optical device is used to detect calcium oxalate crystals and starch grains (amyloplasts). In polarized light, calcium oxalate crystals and starch grains appear as bright, birefringent objects on a dark background. Starch grains observed under polarized light will also have a Maltese-cross effect with the arms of the cross intersecting at the hilum. Calcium oxalate crystals are usually best viewed after the sample has been clarified with Chloral Hydrate Solution or another clarifying agent.
Diluted Acetic Acid—
Add 1 to 2 drops to the plant material, and immediately observe under a microscope: calcium carbonate deposits dissolve with effervescence.
Ferric Chloride Solution—
Dilute 1 mL of
ferric chloride TS with 9 mL of water. For the detection of phenol hydroxyl groups, such as tannins and flavonoids, from the side of the coverslip add the solution to the aqueous sample: tannins and other polyphenols become blue-black to green.
Glycerin—
Use as a mountant to prevent the drying of aqueous and chloral hydrate solutions.
Glycerin–Alcohol Solution—
Mix equal volumes of glycerin and alcohol. Use as a mounting medium.
Glycerin–Gelatin Mixture—
Add 10.0 g of powdered gelatin to 60 mL of water. Allow to stand for 2 hours, and add 70 mL of glycerin containing 1.5 g of dissolved phenol. Heat in a water bath, and filter through a preheated funnel containing glass wool. The filtered mixture is liquefied before use, and it serves as a mounting medium. Add a few drops to the cut or powdered plant material, and cover with a heated coverslip. This preparation is used for long-term storage of specimen mounts. The margins of the coverslip may be sealed with Canada balsam after a few months of drying.
Hydriodic Acid—
Add 1 to 2 drops to plant material: cellulosic cell walls become blue to blue-violet.
Iodine Solution—
Add 1 to 2 drops of 0.1 N iodine VS to the plant material: starch particles become dark-blue to blue-violet; this reaction is reversible on heating. [NOTE—Proteins, lipids, and cellulose turn yellow to brown; and guaiac powder particles become green to blue, but this reagent is not used for diagnostic identification of these features.]
Iodine–Glycerin Solution—
Dissolve 0.3 g of iodine and 1.0 g of potassium iodide in a small quantity of water, and add 10 mL of a mixture of glycerin and water (1:1). Add 1 to 2 drops to the powdered plant material: samples containing saponins form yellow lumps or aggregates. If a sample tests positive for saponin, the result has to be confirmed by testing the sample with Blood–Gelatin Mixture as well.
Lactochloral Solution—
Dissolve 50.0 g of chloral hydrate in 50 mL of lactic acid with gentle heating. Add a few drops to the plant material. Place the microscope slide in a small vacuum desiccator if it is necessary to eliminate air bubbles. Chloral Hydrate Solution and Lactochloral Solution are used for the same type of identification, except that Lactochloral Solution is a stronger clarifying agent and it is used for plant material that is more difficult to clarify.
Lactophenol Solution—
Mix 20 g of lactic acid, 40 g of glycerin, and 20 mL of water. Add 20 g of phenol, and mix. This is a strong clarifying agent suitable for the examination of pollen grains.
Naphthol–Sulfuric Acid Solution—
Prepare a 20% solution of 1-naphthol in alcohol. To plant material add 1 drop of 1-napthol solution and 1 drop of sulfuric acid: inulin crystals turn brownish red and then dissolve.
Osmium Tetroxide Solution—
Dissolve 0.1 g of osmium tetroxide in 5 mL of distilled water. Add 1 to 2 drops of the solution so obtained to plant material: essential oils, fatty oils and other lipids, tannins, and protein bodies become brown to black.
Phloroglucinol–Hydrochloric Acid Solution—
This solution is used for the identification of lignin and other hydroxyphenylpropane derivatives, lignified tissues such as sclereids, vessels, fibers, and stone cells, and lignified parenchyma. Moisten the powder or the cut sample with
phloroglucinol TS, and allow to dry for 2 to 3 minutes before placing the coverslip. Add a few drops of a 25% hydrochloric acid solution, and cover with the coverslip. Lignified cell walls turn carmine red.
[NOTE—This stain is not stable.
] Cells with hydroxyphenylpropane derivatives, such as vanillin and ferulic acid, also turn red. Alternatively, hydroxyphenylpropane derivatives can be extracted from the plant material and the plant material then examined. To extract hydroxyphenylpropane derivatives repeatedly immerse the untreated material in alcohol, mix on a vortex mixer, centrifuge, and discard the alcohol between washings. Then treat the plant material as specified above, beginning with the addition of phloroglucinol TS.
1 M Potassium Hydroxide Solution—
Add 1 drop to plant material: cells containing 1,8-dihydroxyanthraquinones will stain red.
Ruthenium Red Solution—
Add a few drops of ammonium hydroxide to
ruthenium red TS.
[NOTE—Store the solution protected from light.
] Add 1 to 2 drops to plant material: pectin-containing cell membranes, acidic mucilage, and phytoglycogen turn red.
Sodium Hypochlorite Solution—
This solution is used to bleach deeply colored sections. Immerse the plant material in the solution for a few minutes until sufficiently bleached. Wash the tissue with water, and mount with a suitable mounting agent. [NOTE—Sodium hypochlorite will extract lignin; plant tissue so treated will test negative for lignin.]
Sudan III Solution—
Dissolve 0.5 g of Sudan III in 50 mL of alcohol or isopropyl alcohol with reflux boiling. Cool, filter, and add 50 mL of glycerin. Add 1 to 2 drops of this solution to plant powder: essential oils, waxes, cutin, suberin, and fatty oils and other lipids combine with this lipophilic colorant and become orange-red to red after a short time.
Thionine Solution—
Prepare a 0.2% thionine acetate solution in 25 percent alcohol. Immerse the dry sample in this solution. After about 15 minutes, wash out the excess of stain with 25 percent alcohol: mucilage will have swollen into spherical globules and turned red-violet, while cellulose, pectin, and lignified septa will turn blue or blue-violet.
Toluidine Blue Solution—
Using toluidine blue, proceed as directed for Thionine Solution.
Universal Reagent—
SOLUTION A—
Dilute 20 mL of a lactic acid-saturated solution of Sudan III with 30 mL of lactic acid.
SOLUTION B—
Dissolve 0.55 g of aniline sulfate in 35 mL of water.
SOLUTION C—
Dissolve 0.55 g of potassium iodide and 0.05 g of iodine in 5 mL of water, and add 5 mL of alcohol.
PROCEDURE—
Combine Solution A, Solution B, and Solution C, and add 2.5 mL of hydrochloric acid while stirring. [NOTE—The solution is used without filtering.] For identification, add 2 to 3 drops to the sample, and gently boil over a small flame. If necessary, small amounts of Universal Reagent may be added during boiling. Cover with the coverslip: lignified elements turn yellow; suberin turns red-brown; lipids turn red; and starch turns blue-violet.
Water—
Use as a mounting medium. [NOTE—All grades of water are acceptable for this purpose.]
Zinc Chloride–Iodine Solution—
Dissolve 20.0 g of zinc chloride and 6.5 g of potassium iodide in 10.5 mL of water. Add 0.5 g of 0.1 N iodine VS, and shake for 15 minutes. Filter if necessary. Store in low-actinic glassware. Add 1 to 2 drops to plant material, and allow to stand for a few minutes: cellulosic cell walls are stained blue to blue-violet.
Preparation of Temporary Mounts and Hand Sections—
When using the dry plant tissue, soak or gently boil in water until soft. Do not soften too much. Material can then be treated like fresh plant material. When appropriate, use the mountants or reagent solutions listed for use with plant powder to help visualize features of the tissue (see
Preparation and Use of Reagent Solutions,
Optical Devices,
and Mountants).
To make an epidermal peel of the leaf, petal, sepal, bract, and other leaf-like appendages, roll the tissue into a cylinder, and nick with a sharp, polytef-coated razor blade that has been wetted with water. Grasp nicked piece of tissue with forceps, and strip back removing a clear section of the epidermis. Mount in water on a microscope slide, place a coverslip over the tissue, and examine under a microscope. If it is difficult to obtain an epidermal peel using the above procedure, proceed as follows. Soak the tissue in a 40% to 60% nitric acid solution at 60
for 3 to 4 minutes or until the epidermis can be easily peeled. The peel is then washed three to five times in water to remove the excess of nitric acid. Neutralize the tissue in a 1% potassium hydroxide solution or a 1% sodium hydroxide solution. Wash the tissue again with water, mount in water on a microscope slide, place a coverslip over the tissue, and examine under a microscope.
An alternative method of preparing leaf tissue for the examination of the epidermis is to heat a leaf fragment (about 5 mm × 5 mm) for 15 minutes in Chloral Hydrate Solution on a water bath. Transfer the tissue to a microscope slide, add a drop of water, and cover with a coverslip. These procedures can be used to determine the stomatal type, distribution, number, and index.
Stomatal number is determined by counting the number of stomata per unit area of a microscopic field. Determine the stomatal number on at least 10 different sites of the specimen, and calculate a mean value. Keep track of which leaf surface is being observed, abaxial or adaxial, as the stomatal number for different surfaces is frequently significantly different.
To calculate the stomatal index, the specimen is observed under a microscope at a low magnification. The size of the surface is determined with a calibrated micrometer ocular, and the number of stomata and the number of epidermal cells for that area are determined. The stomatal index is calculated by the formula:
100S/(E + S),
in which S is the number of stomata for a given area; and E is the number of epidermal cells of the same area. Determine the stomatal index on at least 10 different sites of the specimen, and calculate a mean value. Again, keep track of which leaf surface is being observed, abaxial or adaxial, as the stomatal indices for different surfaces is frequently significantly different.
To make a cross section of a leaf or thin roots, stems, or other thin appendages, lay the appendage to be sectioned on a microscope slide. Place another microscope slide over the appendage with a portion of the tissue exposed. Using a sharp, polytef-coated razor blade that has been wetted, cut straight down along the edge of upper slide. Without moving the upper slide, cut down again with the razor blade at an angle. Some practice may be necessary for one to be able to get sections thin enough so that when they are mounted and covered with a coverslip, these sections can be used to determine tissue arrangements (for instance, the number of palisade layers in leaf, thickness of cuticle, types of trichomes, types of vascular bundles, and the like). Because razor blades dull quickly, they have to be replaced frequently.
Use the cross section of leaf tissue so obtained to determine the palisade mesophyll ratio. Alternatively, boil leaf fragments of about 2 mm2 in Chloral Hydrate Solution, mount, cover with a coverslip, and observe under a microscope. Identify groups of four adaxial epidermal cells, and count the palisade mesophyll cells that are lying below and are at least 50% covered by the epidermal cells. This value divided by 4 is the palisade mesophyll ratio. Determine the palisade mesophyll ratio of at least 10 groups of epidermal cells, and calculate a mean value. Palisade mesophyll ratio can also be determined on powdered leaf material.
To make a cross section of thick stems, roots, or other plant parts, including woody tissues, hold the tissue in one hand and using a sharp, polytef-coated razor blade that has been wetted with water, shave a cross section from the appendage. Mount in water, another medium, or reagent solution, place a coverslip over the material, and examine under a microscope. Sections thin enough to determine vascular tissue arrangement, ray type, parenchyma distribution, presence of crystals, and the like can usually be made with a little practice.