Imaging—
Images are formed in a SEM system by detection and manipulation of electrons. SE are emitted from a specimen surface as the result of inelastic collisions between primary (incident) electrons (PE) and electrons within a specimen. When the energy imparted to a specimen electron exceeds the work function of a sample, that electron is emitted as an SE. Most SES have energies of 5 to 20 eV; electrons in this low-energy range can be efficiently collected, yielding high signal-to-noise images. Because such low-energy electrons can penetrate only short distances through the specimen, SES originate from within 2 to 30 nm of the surface and generate highly resolved images. The actual PES penetration depth is dependent on PES accelerating voltage, specimen elemental composition, specimen density, and specimen mounting angle. Excitation volumes of 0.5 to 5 µm in diameter are common.
Backscattered electrons are PES that have been reflected from the sample. The PES can undergo multiple collisions prior to exiting from the specimen; therefore, BSES have energies over a broad range and emerge from relatively deep penetration (
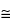
0.1 to 5 µm) (see
Figure 2).
Figure 2. Bulk Penetration.
These high-energy (15 to 25 keV) BSES are collected less efficiently than SES, and they yield images with poorer resolution. The efficiency of BSES reflection is a function of the atomic number (Z) of the specimen atoms; thus, the contrast of BSES images depends on elemental composition. The penetration depth of all electrons is affected by elemental composition, specimen density, specimen tilt, and incident beam energy (accelerating voltage). For example, the SE images of sodium phosphate and zinc phosphate crystals are quite similar. However, the heavier nuclei of the zinc species produce more efficient BSE reflection and BSES images with higher contrast. BSE images of heavy- versus light-element phases, or mixtures of species, show dramatic contrast differences that are representative of elemental heterogeneity.
Although single-angstrom resolution is possible, practical SEM image resolution is limited to
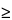
100
(
0.1 µm for X-ray images). These limits depend not only on instrument performance but also on operator acuity. Resolution is optimized under the following conditions: minimum working distance, high accelerating voltage, excellent grounding, excellent mechanical alignment, excellent electronic alignment, minimum incident spot diameter, minimum final aperture diameter, and cleanest column conditions. Sample preparations can be viewed in a variety of orientations and detector modes. Often the examination of a specimen at an oblique angle reveals features unobserved by an electron beam normal to the surface. This is especially true of specimens that have flat, featureless surfaces or that are poor conductors, e.g., glass surfaces. The PE accelerating voltage can be varied to change the PE penetration depth. This procedure is useful for characterizing specimens that are laminated or otherwise heterogeneous between surface and bulk content.
Coating a sample allows observation of a specimen's topography, undisturbed by flare and distortion caused by thermal effects and insufficient grounding. Coatings such as gold, gold–palladium, and carbon are often used because they are highly conductive, easy to apply, and relatively inert. Either evaporation or sputter-coating systems can be used to apply metal films; carbon films must be evaporated. Metal coatings give superior resolution, although their fluorescence can interfere with elemental analysis. Specimen charging affects not only image quality but also X-ray fluorescence yield.
X-ray Emission Analysis—
When a PE encounters an orbital electron in an atom, the resultant collision can either promote that orbital electron to a higher energy level or ionize the atom. Stabilization of an atom by relaxation of a higher energy electron to fill a vacancy results in the emission of an X-ray photon. These X-ray energies are discrete and element-specific; they equal the differences between the shell electron energies for the various shells of a given element. For instance, an ejected
K-shell electron can be stabilized by a higher energy
L-shell electron, yielding a net energy (
EL 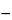 EK
EK), which is specific for the X-ray photon energy of the elemental
K line. X-ray emission lines are classified according to the electron shell in which the vacancy existed, e.g.,
K, L , M. The lines are further categorized according to the shell from which the relaxing electron originates. Thus, a
K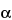
X-ray line arises from a vacancy in a
K-shell that is filled from an
L-shell; a
K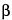
X-ray line arises from a
K-shell vacancy filled from an
M-shell, and so on (see
Figure 3).
Since each shell above
K possesses a number of energy levels, electron transitions yield a number of lines, such as
K1,
K2,
K1, and
K2. The existence of several X-ray emission lines for each element (a few for
Z 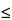
11 and many for
Z 11) is useful in overcoming detection problems due to (1) interelement spectral interferences, e.g., titanium
K and barium
L, (2) sample matrix effects on energy or intensity, and (3) insufficient PE energy to excite some elemental lines, e.g., lead,
K lines.
The energies normally encountered in a SEM/EDX (or WDS) analysis range from 0.28 keV (
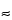
447 nm) for carbon
K to the upper end of the instrument accelerating voltage, typically
40 keV (
1 nm). The natural line width, which is inversely proportional to the lifetime of the upper electronic state, is governed primarily by the transition probabilities for X-ray emission and Auger electron emission. Interaction of X-ray photons with electrons within the specimen can result in Compton scattering to produce a broadened line shifted to lower energy. X-ray photons are also emitted as a result of inelastic acceleration of electrons by atomic nuclei within a specimen. These X-ray photons, termed bremsstrahlung or white radiation, have a broad, continuous energy distribution; and their characteristic lines are superimposed on this background signal.
For lighter elements,
Z 11, the low-energy X-ray photons originating from
K-shell transitions can be detected only with wavelength-dispersive spectrometers or specially configured energy-dispersive detectors. All other elements emit easily detectable X-ray photons. Heavier elements,
Z 16, emit two or more detectable lines corresponding to
K- and
L-shell transitions; and
Z > 57 emit three or more detectable lines corresponding to
K-,
L-, and
M-shell transitions. For a given element, X-ray intensities generally vary as follows:
K >
K >
L >
L, etc. (see
Figure 4).
Figure 4. X-ray Spectrum.
The elemental content of a sample has a bearing on the selection of conditions for analysis. The most useful range of accelerating voltage is
3 to 20 kV; most elements of interest can be ionized by electrons with energies in this range. The energy required in order to excite X-ray emission from a given line is termed its critical excitation potential. The critical excitation potential for a
K line can be approximated by the sum of the primary line energies (
K +
L +
M). Selection of an accelerating voltage equal to 1.5 times this sum is usually sufficient for semiquantitative analyses. For example, copper has
K at 8.05 keV +
L at 0.93 keV = 8.98 keV: 1.5 × 8.98 keV = 13.47 keV. Selection of 15-kV accelerating voltage yields sufficient energy to ionize the
K-shell of copper atoms and generate a useful analytical signal.
Interelement interferences originate from many effects. High-energy X-rays emitted from heavy atoms can ionize lighter elements to produce secondary X-ray emission from the lighter species. Lower high-Z element fluorescence and higher low-Z element fluorescence can be observed, in contrast to that expected from the PE-induced signal of a pure element. Conversely, X-ray emission from a light element may be absorbed by a heavier matrix to yield a negative bias in the light-element signal. These effects always exist in heterogeneous specimens and must be corrected for during any quantitative analysis. A common algorithm, ZAF, may be used to correct for Z-dependent interferences due to absorption and secondary X-ray emission.
;)
;)
;)
;)
;)
;)
;)
;)