PROTEIN CONTENT
Protein content assays are used to quantitatively determine the amount of protein in a given biotechnology-derived product. The determination of protein content is often one of the most difficult measurements that needs to be made and often requires independent confirmation by alternate methods. Where applicable, methods such as UV spectrophotometry with a valid absorptivity and Kjeldahl nitrogen analysis can be used to determine absolute amounts of protein independent of reference standards. However, methods such as Lowry protein, biuret, and quantitative amino acid analysis, which require reference standards, also yield accurate values. Protein content assays are among the most important of all the methods used for these products because the results of other types of assays, such as potency, are also dependent on them.
There are several assays for the determination of protein content that are commonly used. These assays may be used at different points in the production process of a given biotechnology-derived product. For highly pure proteins, the simplest protein content method is based on the determination of the UV absorbance of a protein solution by spectrophotometry. The absorbance at the absorption maximum is determined and the protein concentration is calculated with the use of an empirically determined absorptivity. This technique is applicable to proteins containing the aromatic amino acid residues tryptophan, tyrosine, and/or phenylalanine. The absorption wavelength often used is 280 nm. The extinction coefficient, or molar absorptivity, should be determined in the same solvent that is used for the sample to be measured. If necessary, the product may be diluted prior to analysis to obtain solutions with absorbance values in the linear range of detection. Higher molecular weight aggregates and particulates may give rise to light-scattering effects, which provide artificially high absorbance values. Excipient components that have significant absorbance at 280 nm will also interfere with this test. UV spectrophotometry is unique among the protein content methods in that it is an absolute measure of concentration of a specific protein requiring no calibration with standards.
A commonly used general protein content method is the Lowry assay. This is based on the biuret reaction of proteins with copper (II) in a basic solution and the Folin-Ciocalteu phosphomolybdic-phosphotungstic acid reduction to heteropolymolybdenum blue by the copper-catalyzed oxidation of the aromatic amino acids tyrosine, tryptophan, and phenylalanine in the protein. The reaction products are blue and are quantitated spectrophotometrically in the visible region between 540 and 560 nm. This reaction is linear at microgram protein levels. The assay, however, is prone to interferences from a number of substances such as alcohols, sugars, and detergents. In some cases, interfering substances or product may be removed prior to analysis, e.g., by precipitation. Also, the preparation of controls containing interfering substances that are in the drug product may correct for their presence. Although bovine serum albumin historically has been used to prepare the standard curve, different proteins are known to react with differing intensity, so that a reference material of the same product should be used for calibration. The bicinchoninic acid (BCA) assay is a useful alternative to the Lowry assay because it is less sensitive to interfering substances. The working reagent is a BCA-copper (II) solution. The copper (II) complex is reduced to copper (I) in the presence of protein, and the purple color may be quantitated spectrophotometrically at approximately 560 nm.
Other colorimetric assays can also be used. The Bradford method, for example, employs the binding of the dye Coomassie Brilliant Blue to the protein in an acidic environment. The concentration of the protein in solution is then determined by comparing the absorbance at 595 nm with a standard curve of a reference material.
Fluorescent methods used are normally based on either fluorescamine or o-phthaldialdehyde (OPA). The main advantage of these assays is increased sensitivity. Another advantage is their use with hydrophobic proteins. Fluorescamine and OPA react with primary amines both at the N-terminus of the polypeptide and with amino acid side chains, such as lysine.
The Kjeldahl nitrogen method,
Nitrogen Determination  461
461
, provides an accurate and precise determination of protein concentration and is often used in the determination of UV protein absorptivities. The assay is performed in two stages. The sample is first decomposed with sulfuric acid to produce ammonium sulfate, carbon dioxide, and water. The decomposition is performed at the boiling point of sulfuric acid in long-necked, pear-shaped flasks. These flasks serve to condense water vapor and prevent the loss of material. Depending on the efficiency of decomposition, various salts such as potassium sulfate may be added to increase the boiling point of the sulfuric acid solution. Oxidizing agents such as perchloric acid or potassium permanganate have also been used to improve the decomposition. The second stage of the assay involves the direct determination of ammonia. In most macrodeterminations, ammonia is steam distilled from the mixture after basification with sodium hydroxide. The ammonia can typically be quantitatively distilled out of the mixture in 5 to 20 minutes and absorbed quantitatively into a standardized acidic solution of known volume and normality. The excess acid is then back-titrated with standardized base. For crude determinations of protein, the ammonia value (and therefore the nitrogen content), is multiplied by a factor of 6.25 mg of protein per mg of nitrogen, which corresponds to a nitrogen content of 16%. The protein value so obtained is generally valid for most proteins. If a more accurate value is required, as for an absorptivity determination, then the conversion factor must be calculated for the nitrogen content of the individual pure protein from the known amino acid composition. For glycoproteins that contain amino sugars, the calculated value is biased high unless a correction is applied.
Amino acid analysis is used in the determination of the appropriate absorptivity of the protein and may also be used quantitatively for the determination of protein content. This procedure, although more complicated than those described above, can also yield accurate results.
AMINO ACID ANALYSIS
Amino acid analysis is a classical protein chemistry method for the determination of the amino acid composition of proteins and peptides. The method consists of the complete hydrolysis of a protein or peptide to its component amino acids, which are then chromatographically separated and quantitated. Amino acid analysis, therefore, can be used to determine both the amino acid composition of a product (i.e., identity) and the total amount of protein present. The method has some inherent difficulties, such as complete or partial destruction of some amino acids, that can be circumvented by appropriate analytical methodology. The amino acid tryptophan is destroyed by 6 N hydrochloric acid hydrolysis and thus requires the use of alternate hydrolysis conditions. The amino acids serine and threonine may be partially destroyed, whereas peptide bonds between bulky hydrophobic residues such as valine and isoleucine may be more resistant to hydrolysis, in both cases yielding values lower than actual. Accordingly, analysis of time-course hydrolysis samples may be used to compensate for these factors. Cysteine and methionine may require preoxidation to cysteic acid and methionine sulfone, respectively, for accurate quantitation. Each specific protein may require a procedure of optimized hydrolysis conditions for its amino acid analysis to obtain the optimal results.
Amino acid analysis is performed in two stages. The first stage involves the hydrolysis of the protein into its component amino acids. This hydrolysis is normally performed with 6 N hydrochloric acid at about 110
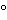
for 24 hours. Some proteins may require longer or more stringent hydrolysis conditions. The second stage is the separation and quantitation of the individual amino acids by some form of chromatography that can be performed with either precolumn or postcolumn derivatization. A number of precolumn derivative procedures are available, such as with OPA, phenylisothiocyanate (PITC), and fluorenylmethoxycarbonyl (FMOC). These derivatives are then separated by reversed-phase (RP) high-performance liquid chromatography (HPLC) and quantitated following UV or fluorescence detection. Postcolumn derivative methods involve separation of the component amino acids by high-performance ion-exchange chromatography (HPIEC) followed by postcolumn reaction with a chromophore, such as ninhydrin, and quantitation following UV/visible detection. All of these methods are suitable for performing amino acid analyses and each has its inherent advantages and disadvantages. OPA derivatives are very simple to prepare and are sensitive, requiring only a small amount of sample, but they are unstable and have to be chromatographed immediately upon preparation. Phenylthiocarbamyl (PTC) derivatives, on the other hand, are relatively more stable. Postcolumn derivatization with ninhydrin is often performed in the low-pressure mode and has the advantage of stability of the amino acid hydrolysate. Its disadvantage is the need for dual detection at 440 and 570 nm and for post-column apparatus.